


Gut bacteria in the blood of COVID-hospitalized patients?
A study provides a possible mechanism of gut translocation and bacteremia in SARS-COV2 patients, however with some serious limitations and possible alternative explanations.

tl;dr: This article was an assessment of what I assumed to be a bombshell article, tying gut translocation to sepsis and providing an explanation for the ARDS witnessed in hospitalized COVID patients. However, after spending a good deal of time looking at the study I realized that it wasn’t what I thought it was upon first glance. The proceeding article is an assessment of the study, providing additional context for some of its findings, as well as pointing out some issues with the study and why it’s not the bombshell article it appeared to be. Look for Key Takeaways for a quick rundown of the findings. A proper conclusion and hypothetical model will be provided in a following article in the near future.
So far we’ve discussed different aspects of the microbiome as it relates to viral infection:
Viral infections apparently have an influence on the microbiome. Although direct infection and injury is a possible explanation, it’s been assumed that systemic inflammation during a viral infection likely plays a role in gut dysbiosis.
Bacteria may translocate from the gut to the lungs during sepsis and ARDS. Gut dysbiosis has been associated with sepsis, although the actual relationship between the two is unknown. We took a look at one study (Dickson, et al.1), in which some anaerobic bacteria were found in the lungs of ARDS patients. this would infer a possible movement of gut bacteria to the lungs, although this was not studied within the cited Dickson, et al. report.
It’s also known that SARS-COV2 can lead to gut dysbiosis, and SARS-COV2 in severe cases may lead to sepsis and ARDS, of which the current cause of the disease is not yet known.
Given all of this information, one may hypothesize that sepsis and ARDS in SARS-COV2 patients may be related to gut dysbiosis.
So far no information has been published to provide an association between this translocation and sepsis in COVID patients.
However, an article was published in November 2022 that may help shed some light and piece a few things together. This study looked at patients hospitalized for COVID and noted that several patients had evidence of bacteremia (bacteria in the blood), and that some of these bacteria were also found in the gut of the corresponding patients, suggesting that bacterial translocation may have caused bacteria to move from the gut to the blood during the course of a SARS-COV2 infection.
We’ll dive into the study, while also noting some of the issues I take with it, as well as why the hospital may play a factor in ARDS.
Relevant Terminology
alpha diversity: This term is used often within the proceeding study, as well as many microbiome studies. It’s an ecology term that describes the diversity of species within a given environment. In this case, alpha diversity refers to the strains of bacteria that make up a healthy gut. Its use in this study will generally come in the phrase “decrease/decline in alpha diversity”, and it just means that the bacteria in the gut of SARS-COV2 patients became less diverse relative to healthy individuals (i.e. fewer bacterial species were found in SARS-COV2 patients).
Bloodstream Infection (BSI): A general term used to explain the presence of infectious agents within the bloodstream. A more specific example would be bacteremia, in which case the agents within the bloodstream are bacterial (viremia for viruses and fungemia for fungi and yeast). The presence of these agents may cause sepsis and a dangerous inflammatory response. BSI and sepsis are some of the top reasons for mortality in hospitalized patients.
Note that the researchers may refer to BSI with the letter “n” as in nBSI, which denotes nosocomial, or hospital-acquired infection.
Goblet cells: Goblet cells are a special type of gut epithelial cell involved with the production of the mucous agent mucin. Their activity is necessary in order to maintain the mucosal lining of membranes and epithelium, as a decrease in their activity may lead to permeability. In the case of the gut, lack of mucin may cause gut permeability (akin to leaky gut or IBS) and aid in movement of bacteria from the gut into the blood.
Paneth cells: Highly specialized secretory epithelial cells found within the small intestines. Their roles are multifaceted, and are usually involved with maintaining the homeostasis of the small intestines. They also secret antimicrobial agents and are involved with immune responses to protect the gut from pathogens. Therefore, reduced activity may be associated with susceptibility to pathogens and infections.
Plaque-forming units (PFUs): You may have seen PFUs when looking at SARS-COV2 or other viruses. It’s a proxy measure to explain how many plaques the virions within a solution can form. Plaques are the clear regions that can be seen on a petri dish lined with a monolayer of cells. Plaque formation is an indication that the cells from that monolayer were lysed, thus leaving empty circles on the petri dish.
A solution that makes mention of 1000 PFUs/ mL would suggest that there’s approximate enough virions within one milliliter of a solution to form 1,000 plaques.
The proceeding study challenged mice with 10,000 PFUs, which would infer enough virions to create 10,000 plaques.
A look at the study
The study in question comes from Bernard-Raichon, et al.2 published in Nature on November of 2022:

Upon first glance the mention of antibiotic-treated patients may raise some concern because there is clearly another variable being looked at. However, given that nearly all patients were prescribed antibiotics upon admission to hospitals for COVID (the rule rather than the exception) this may be an acceptable loss. This will also be something worth mentioning later on, as it will provide some additional context.
Introduction
I normally don’t include excerpts from the Introduction section, however I think it provides some background information, with the researchers noting a few things I have already noted throughout the previous posts:
Complex gut microbiota ecosystems can prevent the invasion of potentially pathogenic bacteria15,16. Conversely, when the gut microbiota incurs damage, such as through antibiotics treatment, competitive exclusion of pathogens is weakened17–19 and potentially dangerous blooms of antibiotic-resistant bacterial strains can occur20,21. In immunocompromised cancer patients, blooms of Enterococcaceae and Gram-negative proteobacteria can lead to gut dominations by few or single species22–25. Such gut domination events are dangerous to these patients because they are associated with increased risk of translocation of antibiotic-resistant bacteria from the gut into the blood stream22,26,27. Bacterial co-infection can also cause life-threatening complications in patients with severe viral infections10,11,28; therefore, antibacterial agents were administered empirically to nearly all critically ill suspected COVID-19 patients since the incidence of bacterial superinfection was unknown early during the pandemic4,29. However, it is now known that nosocomial infection during prolonged hospitalization is the primary threat to patients with COVID-1930, rather than bacterial co-infection upon hospital admission12,31–33. Evidence from immunocompromised cancer patients suggests that indiscriminate administration of broad-spectrum antibiotics may, counter-intuitively, increase nosocomial bloodstream infection (nBSI) rates by causing gut dominations of resistant microbes that can translocate into the blood22,34. Indeed, we recently showed that Enterococcus, a common gut microbial genus comprised of intrinsically antibiotic-resistant strains, accounts for a large proportion of nBSIs during longer hospitalizations, suggesting gut translocation35. Thus, empiric antimicrobial use, i.e., without direct evidence for a bacterial infection, in patients with severe COVID-19 may be especially pernicious because it can select for antimicrobial resistance and promote gut translocation-associated nBSI.
In short, evidence now suggests that a good deal of hospitalized COVID patients have been recognized as having bacterial co-infections, and it’s possible that secondary bacterial infections may contribute to severity of COVID, possibly through sepsis.
Note the researchers comment on the use of antibiotics in influencing gut translocation here, and that there may be some concern that the attempts at sterility within a hospital may also make way for antibiotic-resistant bacteria to infect patients (refer to my post on the sterility bubble for comments on the hospital).
This idea will, again, be something worth considering further along.
Mice Results
Similar to the Dickson, et al. study the researchers here used mice as a reference model for humans. Note that the intent of using mice models is to see what researchers should look for in human models i.e. are there certain bacterial strains that gain dominance in SARS-COV2-infected mice that should be looked for in infected humans?
Of course, remember that humans aren’t mice, even if humans may act rodent-like at times. This comes with the caveat that mice may have different microbiomes compared to humans, and that laboratory mice in particular are going to be subject to controlled microbial exposure that would also influence their microbiome as well.
In this experiment, researchers challenged mice with 10,000 PFUs3 intranasally of SARS-COV2 and compared their microbiome daily to those of control mice by examining their feces with genomic sequencing. Mice were sacrificed around Day 6 for gut analysis.
Microbiome alterations in SARS-COV2 infected mice
Be forewarned that the following figure may seem convoluted at first, but is actually easy once broken down. Note that the researchers provide a closer look at the 10,000 PFU cohort in the Supplementary Material, which can be found in the footnotes4 of this article.
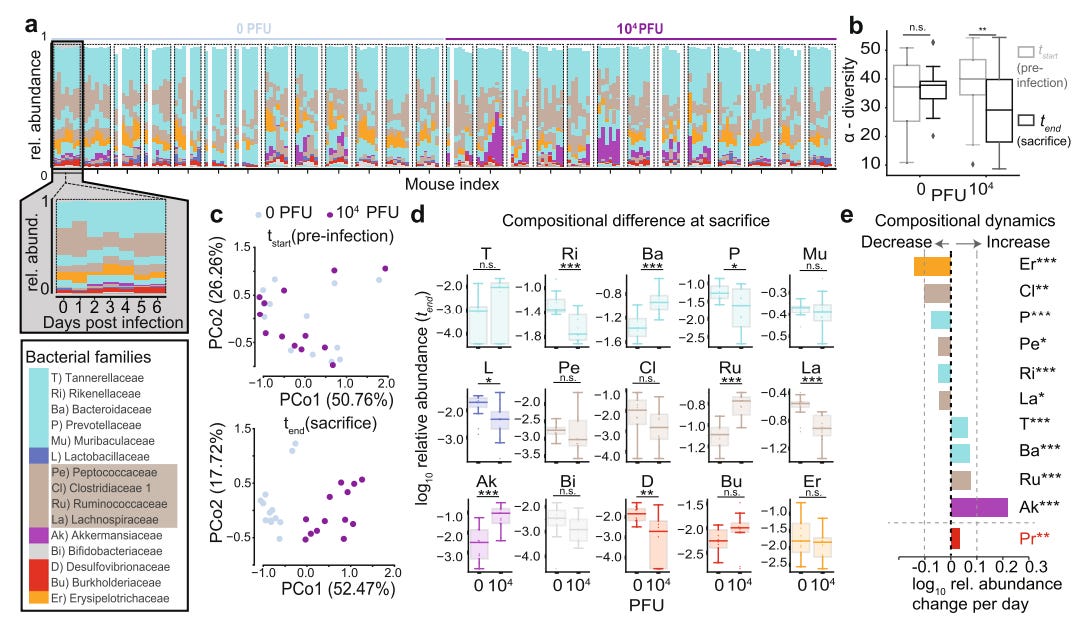
First off, there’s a lot of color on the legends and some of the graphs above. Note that everything is intentionally color-coded in the same manner i.e. the bacterial family Akkermansiaceae (AK) is in purple in a,d, and e.
The first part of the figure (Fig. 1a) compares the abundance of various bacterial families in both control mice and the infected mice, color-coded to indicate which families were in high abundance relative to other ones. Each rectangle represents one mouse within a given cohort.
Note the expanded box in grey which provides a closer view of the breakdown over the course of 7 days, with Day 0 serving as a baseline before challenging with SARS-COV2. What’s significant is not where each bar lies in the graph, but how large the bar is and whether it changes over the course of the week, which would indicate alterations in abundance of that particular family.
It also becomes more clear when looking at both Fig. 1d and Fig. 1e, with the former showing differences in relative abundance of bacterial families between control and infected mice at Day 6, and the latter showing average daily changes.
Altogether, it’s apparent that alpha diversity was lower in infected mice by Day 6 when compared to control mice (Fig. 1b).
In particular, significant increases were seen among bacteria within the Akkermansiaceae, Ruminococcaceae, and Bacteroidacea families, a few of which are part of the Proteobacteria phylum and are associated with intestinal diseases5. In contrast, there were significant declines in bacteria from the Rikenellaceae and Lachnospiraceae families.
Similar to prior articles, it’s not significant to know every family (note family is used in this figure), however it’s interesting to note that Bacteroidacea were increased in these mice models, as the Dickson, et al. study noted increases in Bacteroides (the genus) in ARDS patients.
Granted, not all Bacteroides are associated with COVID severity, as several were considered to be protective as noted in the Zuo, et al.6 study, possibly due to their downregulation of ACEII receptors within the gut. So the use of families here is very broad and doesn’t tell which species may be the concerning agents.
Also, Akkermansiaceae will be given some special attention further down.
Unfortunately, if you look at the relative abundance for the control mice over the one week period, you’ll note that many mice had different relative abundances compared to one another, and that several appear to change over time as well, as can be seen in mice 5, 8, 9, and 10 (from left to right).
The researchers don’t note daily changes in relative abundance for the control mice similar to Fig. 1e for infected mice, and they also don’t prove baseline comparisons between control and infected mice, which would help with Fig. 1d as it would note whether both groups experienced a change in relative abundance over time as well.
SARS-COV2 infection altered gut epithelia of mice
Next, the researchers compared the ileum (end of the small intestine) of control mice and infected mice. The intent here is to look for morphological changes in the gut that may be related to possible translocation of bacteria, possibly by noting factors associated with gut permeability.
Here, the researchers noted an increase in goblet cells and a decrease in Paneth cells, with the decrease in Paneth cells leading to a decrease in antimicrobial expression of infected mice.
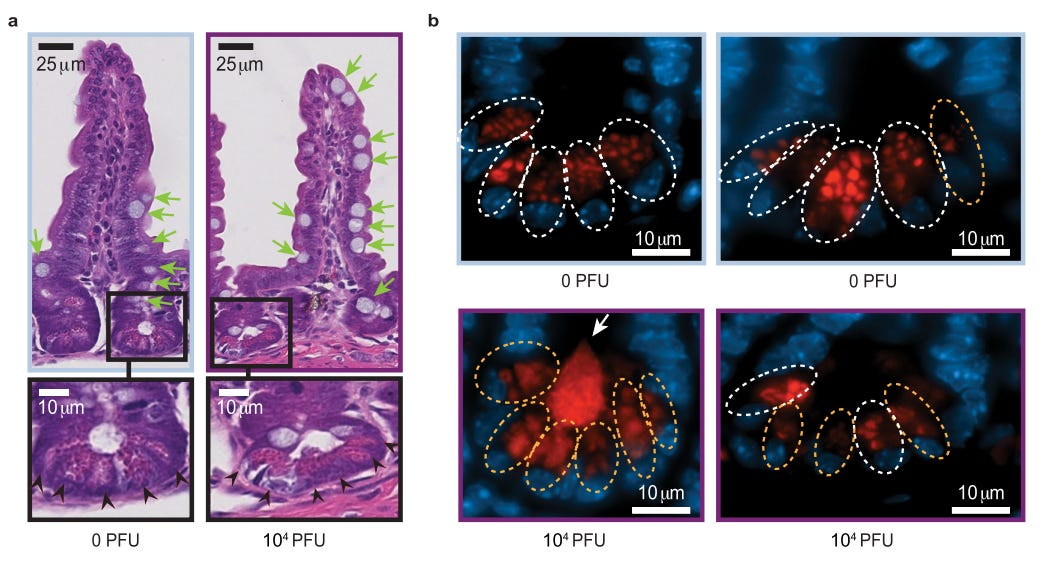
Green arrows in Fig. 2a denote goblet cells, and it appears that goblet cell expression was elevated in infected mice relative to control mice. Black arrows note Paneth cell expression as made clear in Fig. 2b.
Here, the lysozymes (antimicrobial enzymes) of Paneth cells were stained red to examine their patterns. White dotted circles noted normal Paneth cells while yellow dotted circles noted abnormal cells.
From this, it appears that higher abnormal lysozyme expression was noted in the Paneth cells of infected mice. However, note that the use of “abnormal” here is both subjective and ambiguous, as the image on the right for 10,000 PFU shows reduced lysozyme expression while the left image shows elevated expression.
The researchers describe “abnormal” within the Fig. 2b’s caption with the following:
Abnormality is characterized by distorted, depleted, or diffuse lysozyme distribution patterns in Paneth cells.
Again, subjective and ambiguous, as it doesn’t explain the differences between the two images (are some Paneth cells overexpressing lysozymes in order to combat pathogenic agents, or are signaling mechanisms leading to downregulation of lysozyme expression in some Paneth cells, possibly tied to inflammatory markers and SARS-COV2?).
When comparing goblet cells and abnormal Paneth cells to alpha diversity, the researchers noted a negative correlation between alpha diversity and these two cells. Essentially, higher goblet cells and abnormal Paneth cells were associated with reduced alpha diversity (Fig. 2c, not shown).
In particular, the researchers wanted to see if there was a correlation between relative Akkermansiaceae abundance and quantity of goblet cells and abnormal Paneth cells. Interestingly, there appeared to be a strong correlation between the variables:
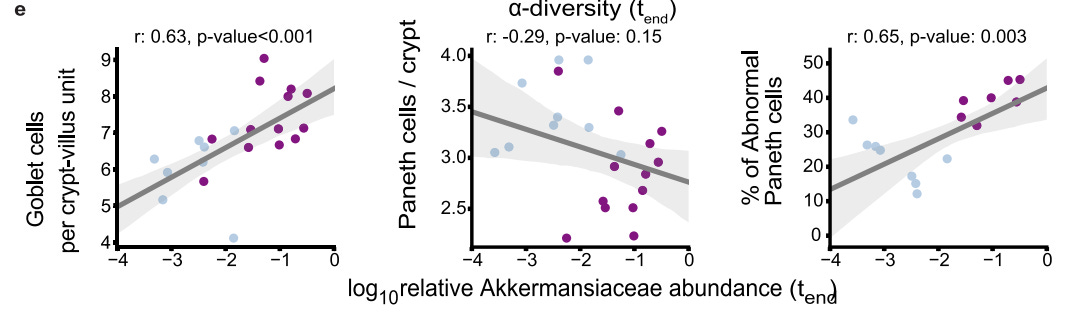
It’s worth noting that the number of Paneth cells didn’t appear to significantly correlate with relative Akkermansiaceae abundance, which may suggest cellular dysfunction of Paneth cells as being a variable rather than quantity of Paneth cells.
So why examine Akkermansiaceae in particular?
Aside from several mice showing elevated abundance of Akkermansiaceae, such as mice 2-5 from Fig. 1a, species from the Akkermansiaceae family are considered to be opportunistic bacteria.
The most widely studied species is Akkermansia muciniphila. As its name suggests, this species loves the mucin that comprise mucous membranes and is involved in the breakdown.
In this study it’s interesting that goblet cell numbers were correlated with Akkermansiaceae abundance, as goblet cells produce the mucin which Akkermansiaceae break down.
Therefore, it’s possible that the goblet cell numbers may be elevated as a response to mucin degradation due to elevated Akkermansiaceae levels.
However, the elevated abundance of Akkermansiaceae would have to be contextualized. In recent years Akkermansiaceae have become a probiotic of interest due to their role in tumor immunotherapy and metabolic diseases such as diabetes and obesity. Their presence during cancer treatment has been known to increase responsiveness to treatments in patients7, suggesting an immunoregulatory mechanism. They’ve also been noted to be elevated in cancer patients as well. However, their role in irritable bowel disease (IBD) has been one of controversy, as several studies have noted both an anti-inflammatory and pro-inflammatory mechanism for this species8.
So why exactly Akkermansiaceae increased here is left up to speculation, as it could be either secondary to the bacterial infection or it could itself act as a pathogenic agent by removing mucin and increase gut permeability.
Again, the fact that the rise of Akkermansiaceae abundance among mice was inconsistent, and given the actual role of this bacteria in gut permeability, and the missing link between SARS-COV2 infection and the abundance raises several questions as to what the abundance of Akkermansiaceae may indicate with respect to SARS-COV2.
The researchers do note that the more sick mice showed a greater decline in biodiversity, however they don’t note if this pertains specifically to the abundance of Akkermansiaceae.
The researchers do make this association within the text of their article:
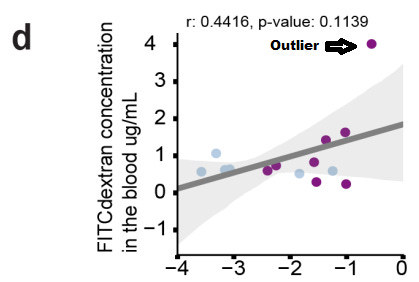
Interestingly, infected mice that had incurred the most severe microbiome injury in the form of diversity loss also showed the most evidence of gut permeability—the highest FITC-dextran concentrations in the blood of mice detected across all samples came from the mice with the most extreme dysbiosis and highest levels of Akkermansiaceae, a family of mucin-degrading bacterial species (Supplementary Fig. 4).
FITC-dextran (Fluorescein isothiocyanate–dextran) is a compound used to measure cell permeability such as in blood-brain barrier permeability studies, so the elevated concentrations of FITC-dextran may suggest some permeability, although the correlation here didn’t appear to meet statistical significance and only appeared in one possible outlier.9
Mice Key Takeaways
Altogether, what exactly do the mice studies highlight?
They show that SARS-COV2 infection clearly leads to microbiome alterations, something noted in several studies as being a rather common feature of viral infections.
However, with respect to the microbiome the results here are rather inconsistent, which is ironic as the lower PFU-infected mice were not used due to their inconsistent results and lack of symptoms (noted in Supplementary Fig. 2).
In a few mice mice there were noticeable increases in several bacterial families such as the Akkermansiaceae and Bacteroidacea families, as well as a decrease in other commensal families, although the actual relationship between these alterations and gut permeability are rather ambiguous.
The researchers single out the Akkermansiaceae family for their role in deterioration of the mucous lining of gut epithelial—something that would likely influence gut permeability. However, the results provide proxy information, and the actual study that served as a model for gut permeability via the use of FITC-dextran did not note significant differences in permeability between control and infected mice.
As such, the evidence here points to microbiome dysbiosis, but doesn’t provide much in the way of providing evidence of gut permeability, which is made even more strange given that the researchers did not conduct sequencing for the possibility of bacteremia. However, this may be due to the timing of euthanizing mice which may not have allowed for bacterial translocation into the bloodstream.
That leaves us with the human studies, and it’s something I had to look at for quite some time to figure out what exactly is going on.
Clinical Study Results
To examine bacteremia in humans researchers looked at hospitalized COVID patients from NYU Langone Heath (n=60 patients) and Yale New Haven Hospital (n=36 patients). A total of 130 fecal samples were taken from patients throughout the study period, with multiple samples taken longitudinally if possible.
Note that these patients were prescribed antibiotics, and so microbiome alteration would need to be looked at within the context of antibiotics as being a contributory mechanism.
For our interests, we’ll take a look at the BSI patients in particular as they are the aspect of this study that is worth examining.
We’ve already looked at gut dysbiosis in SARS-COV2 patients in a prior article, so this section will be skipped.
But for those interested note that Fig. 3 jumps from using bacterial family for Fig. 3a to phyla in Fig. 3d to genus in Fig. 3i, so don’t get confused with how these are named.
Also, note the red dots in Fig. 3a, which indicate an abundance of over 50% for one bacterial family and are associated with reduced alpha diversity (a bit of a redundant statement in some regard).
Members of the Faecalibacterium genus appeared to have also declined the most in these patients (Fig. 3i). Species from the Faecalibacterium genus of bacteria are some of the most critical to our overall health10, as they are involved with the fermentation of fiber to produce short chain fatty acids; anti-inflammatory molecules. Here, there’s an apparent correlation between Faecalibacterium decline and increased risk of BSI.
With that out of the way, the researchers noted that all but 6 BSI patients (25 in total:15 from NYU and 10 from Yale) were prescribed antibiotics prior to BSI detection (check footnotes11 for the breakdown of antibiotic use and sample collection). This may raise some ideas as to whether the use of antibiotics may have contributed to BSI through removal of protective bacteria.
Now, this brings us to the results that I have looked at extensively in order to make sense of this information.
Of course, when researchers are noting BSI, it would be imperative to compare the bacteria from blood samples with stool samples to identify similarities.
To that, the researchers make this comment:
We therefore next investigated a direct association between the bacteria populating the gut microbiome and the organisms identified in the blood of patients. Visualizing the bacterial composition in stool samples from patients alongside the BSI microorganisms (Supplementary Fig. 9a) suggested a correspondence with the respective taxa identified in the blood: high abundances of the BSI-causing microbes were found in corresponding stool samples. A rank abundance analysis matching the organisms identified in clinical blood cultures to the composition of bacteria in corresponding stool samples indicated enrichment of taxa belonging to the same bacterial orders as BSIcausing organisms (Supplementary Fig. 9b), suggesting translocation of bacteria from the gut into the bloodstream.
Strangely, this information isn’t included in the actual study and was relegated to “Supplementary Figure status”. In fact, many of the results for the BSI patients were moved to the Supplementary Figures, and upon looking at the information there’s a bit here that’s worth discussing.
The proceeding graph compares genomic sequencing data between bacteria from the blood with corresponding stool samples for BSI patients:

You’ll notice that many of the bacteria found within the blood were Staphylococcus, denoted with bright yellow bars. However, if you look at the stool samples you’ll notice that not many have that corresponding yellow bar.
Now, this comparison may not be one-to-one, as some other bacteria found in the blood were not seen in the stool, such as the samples that contained Proteus and one with Serratia. Also, even a bar noting a 0.1% abundance of a bacterium may not be visually noticed on the above chart but may still be part of the gut microbiome and recognized through sequencing.
With that being said, the high overall prevalence of Staphylococcus was still worth looking into. But here, the researchers actually provided a possible explanation for what species the Staphylococcus bacteria were of.
In order to identify the bacteria the researchers conducted shotgun metagenomic sequencing and received the following results, with Staphylococci species boxed in black:

Note that many of the identified Staphylococci species are of the species Staphylococcus epidermidis and Staphylococcus hominis, species found predominately on the skin of humans. S. epidermidis is noted to also colonize mucous membranes, but S. hominis in particular is a skin microbiome that plays a role in our body odor.
And so the fact that many of these skin microbes were identified within the blood of BSI patients raised some suspicions.
Of course, the first assumption would suggest that the puncturing of skin during blood collection may have contaminated the samples, which is a possibility in this scenario. Clinicians tend to face these issues when assessing blood samples.
But there’s also the possibility that hospital interventions may also contribute to bacteremia as nosocomial infections. Intravenous catheters, mechanical ventilation and intubation, as well as other invasive interventions may allow these bacteria a pathway into the bloodstream and infect patients.
Note that staph infections are some of the top hospital-acquired infections, with both S. epidermidis12 and S. hominis13 in particular being rather common.
The researchers don’t note much with respect to whether these BSIs may be related to nosocomial infections, or if they may be a consequence of gut microbe translocation, but that doesn’t mean this is not worth considering.
Many Staphylococci species are prone to being antibiotic resistant, with one of the most common antibiotic-resistant subspecies of S. hominis being attributed to many nosocomial infections. Remember that many of these BSI patients were treated with antibiotics, as was common for many SARS-COV2 patients during the early stages of the pandemic, and possibly even to this day.
It’s worth noting that a higher number of BSI patients had pneumonia, had sepsis, had encephalopathy, were intubated, and/or were admitted to the hospital for far longer relative to non-BSI patients.14
Altogether, it's quite possible that the Staphylococci noted within the blood of BSI patients may have been derived externally whether through the hospital or just through the skin of patients.
The researchers make a comment with respect to this possibility in the Discussion:
We presented evidence that microorganisms from the dysbiotic gut microbiome translocate into the blood of COVID-19 patients, plausibly due to a combination of the immunocompromising effects of the viral infection and antibiotic-driven depletion of commensal gut microbes. However, COVID-19 patients are also uniquely exposed to other potential factors predisposing them to bacteremia, including immunosuppressive drugs, long hospital stays, and catheters and our study is limited in its ability to investigate their individual effects.
But this also doesn’t mean that gut translocation is not a viable factor here.
When comparing the other BSI samples without noted Staphylococci present the researchers suggested that these species possibly came from the gut:
In all investigated cases of positive blood cultures by organisms other than Staphylococcus, the species identified in clinical blood cultures had corresponding reads in the stool samples. Strikingly, the relative abundances of matched species tended to be larger than the average abundances of matched species across all samples (Supplementary Table 3).
Note that species such as B. thetaiotaomicron, E. coli, and the Lactobaciullus species are all gut bacteria, with the first two being common opportunistic pathogens, and may serve as actual indications of translocation into the blood of these patients.
Clinical Key Takeaways
This section ended up taking up quite a bit of time to analyze due to some of the strange results (quite some time being an understatement…).
Upon first glance, this would seem like a good study to tie together a possible relationship between gut translocation during SARS-COV2 and ARDS by noting the movement of gut bacteria into the bloodstream of severe patients.
However, the evidence is far too muddy to make such an assertion.
Typical of many human microbiome studies there is no ideal microbiome to reference. As such, many patients are likely to have wide differences in gut flora. Compounded with SARS-COV2 infection, timing of sampling, and the use of antibiotics it becomes hard to consider only one variable alone.
Because of this, it’s hard to argue exactly what the microbiome should look like for these patients without a prior baseline to compare.
However, when comparing the bacteria from blood samples to fecal bacteria there does appear to be some similarities, which may suggest movement of gut bacteria into the blood of BSI patients.
How these bacteria moved is not clear. The researchers suggest that gut permeability may be the reason for this gut translocation, but as noted previously other factors such as medical interventions may be responsible for the bacteremia. The results from the mouse models note alterations of defensins, which are antimicrobial agents. It’s been suggested that alterations to defensins may be associated with inflammation and may play a role in gut permeability akin to IBD.
Indeed, it’s possible that gut permeability and microbe translocation may be a consequence of something as ubiquitous as inflammation driven by the viral infection alone.
Interestingly, the researchers noted a drastic reduction in bacteria from the Faecalibacterium family, a key bacteria critical for its anti-inflammatory properties through production of short-chain fatty acids. Prior studies with antibiotic-naïve SARS-COV2 patients also noted a decrease in Faecalibacterium, suggesting a viral/inflammatory mechanism as the cause rather than antibiotic use.15
Altogether, the study notes that gut bacteria were present within the blood of some patients, but not exactly how this gut translocation occurred. Evidence may point to inflammation being the key driver, as noted in another study in which plasma markers for gut permeability and inflammation were elevated in severe COVID patients16.
The next article will provide a proper conclusion, as well as a hypothetical model for sepsis and ARDS, again with many caveats.
If you enjoyed this post and other works please consider supporting me through a paid Substack subscription or through my Ko-fi. Any bit helps, and it encourages independent creators and journalists outside the mainstream.

Dickson, R. P., Singer, B. H., Newstead, M. W., Falkowski, N. R., Erb-Downward, J. R., Standiford, T. J., & Huffnagle, G. B. (2016). Enrichment of the lung microbiome with gut bacteria in sepsis and the acute respiratory distress syndrome. Nature microbiology, 1(10), 16113. https://doi.org/10.1038/nmicrobiol.2016.113
Bernard-Raichon, L., Venzon, M., Klein, J., Axelrad, J. E., Zhang, C., Sullivan, A. P., Hussey, G. A., Casanovas-Massana, A., Noval, M. G., Valero-Jimenez, A. M., Gago, J., Putzel, G., Pironti, A., Wilder, E., Yale IMPACT Research Team, Thorpe, L. E., Littman, D. R., Dittmann, M., Stapleford, K. A., Shopsin, B., … Schluter, J. (2022). Gut microbiome dysbiosis in antibiotic-treated COVID-19 patients is associated with microbial translocation and bacteremia. Nature communications, 13(1), 5926. https://doi.org/10.1038/s41467-022-33395-6
When comparing lower doses the researchers noted that the dysbiosis was heavily uneven, with many mice not showing any symptoms, and so the higher dose was used.

Rizzatti, G., Lopetuso, L. R., Gibiino, G., Binda, C., & Gasbarrini, A. (2017). Proteobacteria: A Common Factor in Human Diseases. BioMed research international, 2017, 9351507. https://doi.org/10.1155/2017/9351507
Zuo, T., Zhang, F., Lui, G. C. Y., Yeoh, Y. K., Li, A. Y. L., Zhan, H., Wan, Y., Chung, A. C. K., Cheung, C. P., Chen, N., Lai, C. K. C., Chen, Z., Tso, E. Y. K., Fung, K. S. C., Chan, V., Ling, L., Joynt, G., Hui, D. S. C., Chan, F. K. L., Chan, P. K. S., … Ng, S. C. (2020). Alterations in Gut Microbiota of Patients With COVID-19 During Time of Hospitalization. Gastroenterology, 159(3), 944–955.e8. https://doi.org/10.1053/j.gastro.2020.05.048
Grenda, A., Iwan, E., Chmielewska, I., Krawczyk, P., Giza, A., Bomba, A., Frąk, M., Rolska, A., Szczyrek, M., Kieszko, R., Kucharczyk, T., Jarosz, B., Wasyl, D., & Milanowski, J. (2022). Presence of Akkermansiaceae in gut microbiome and immunotherapy effectiveness in patients with advanced non-small cell lung cancer. AMB Express, 12(1), 86. https://doi.org/10.1186/s13568-022-01428-4
Zhang, T., Ji, X., Lu, G., & Zhang, F. (2021). The potential of Akkermansia muciniphila in inflammatory bowel disease. Applied microbiology and biotechnology, 105(14-15), 5785–5794. https://doi.org/10.1007/s00253-021-11453-1
From Supplementary Fig. 4d. when FITC-dextran concentration in blood was plotted against Akkermansiaceae abundance one outlier was noted, which may have driven the positive correlation here. Note blue dots are control mice and purple dots are infected mice. Irrespective of this outlier, the results still don’t meet statistical significance. Although not the end-all, be-all of proper results, it at least adds to the fact that these results may be conflated by the outlier.
Lopez-Siles, M., Duncan, S. H., Garcia-Gil, L. J., & Martinez-Medina, M. (2017). Faecalibacterium prausnitzii: from microbiology to diagnostics and prognostics. The ISME journal, 11(4), 841–852. https://doi.org/10.1038/ismej.2016.176
The following chart from Supplementary Fig. 6 outlines the timing of antibiotics, stool sample collection, and BSI detection with respect to positive COVID test:

Ziebuhr, W., Hennig, S., Eckart, M., Kränzler, H., Batzilla, C., & Kozitskaya, S. (2006). Nosocomial infections by Staphylococcus epidermidis: how a commensal bacterium turns into a pathogen. International journal of antimicrobial agents, 28 Suppl 1, S14–S20. https://doi.org/10.1016/j.ijantimicag.2006.05.012
Kloos, W. E., George, C. G., Olgiate, J. S., Van Pelt, L., McKinnon, M. L., Zimmer, B. L., Muller, E., Weinstein, M. P., & Mirrett, S. (1998). Staphylococcus hominis subsp. novobiosepticus subsp. nov., a novel trehalose- and N-acetyl-D-glucosamine-negative, novobiocin- and multiple-antibiotic-resistant subspecies isolated from human blood cultures. International journal of systematic bacteriology, 48 Pt 3, 799–812. https://doi.org/10.1099/00207713-48-3-799
**Note that this is relevant if the S. hominis species found in the blood are of the subspecies novobiosepticus, which is considered to be highly resistant to antimicrobials.
From Supplementary Table 2. Keep in mind that the sample sizes are not the same with BSI patients having nearly half the population relative to non-BSI. Also, the “26” may be a typo as the rest of the study mentions there being 25 BSI patients.

Hazan, S., Stollman, N., Bozkurt, H. S., Dave, S., Papoutsis, A. J., Daniels, J., Barrows, B. D., Quigley, E. M., & Borody, T. J. (2022). Lost microbes of COVID-19: Bifidobacterium, Faecalibacterium depletion and decreased microbiome diversity associated with SARS-CoV-2 infection severity. BMJ open gastroenterology, 9(1), e000871. https://doi.org/10.1136/bmjgast-2022-000871
Giron, L. B., Dweep, H., Yin, X., Wang, H., Damra, M., Goldman, A. R., Gorman, N., Palmer, C. S., Tang, H. Y., Shaikh, M. W., Forsyth, C. B., Balk, R. A., Zilberstein, N. F., Liu, Q., Kossenkov, A., Keshavarzian, A., Landay, A., & Abdel-Mohsen, M. (2021). Plasma Markers of Disrupted Gut Permeability in Severe COVID-19 Patients. Frontiers in immunology, 12, 686240. https://doi.org/10.3389/fimmu.2021.686240
"However, given that nearly all patients were prescribed antibiotics upon admission to hospitals for COVID" this is my impression as well, but do you have a source for how often antibiotics were actually used?
That antibiotics were withheld has become a new conspiracy theory thanks to jikky/arkmedic https://arkmedic.substack.com/i/79557186/what-does-matter-then But I see several studies reporting high use for hospitalized patients in discrete settings so I think the conspiracy theory is a whiff.
A weak gut barrier Seems to be the common explanation among alternative health practitioners (checkout the healthmeans summits) for many ailments. healing the gut seems to be highly recommended as a source of health. Intermittent fasting allows healing time. the lining can get microscopic tears (leaky) from exposure to food containing the chelator-glyphosate, toxins such as toxic proteins, metals and undigested stuff and of course medications.