

Correction: As Brian Mowrey mentioned in the comments in my previous article note that the reference to the cells in this study are “leukocytes”. This is likely due to the cells that are being sourced, as blood samples were taken from patients. Note that my explanation is simplistic as it doesn’t take into account that the results are specific to leukocytes, as in we can’t infer what is happening in other cells. It also has to take into account whether the sample leukocytes are representative of all of the patients’ leukocytes as well. Because of these factors it isn’t correct to try and compare the allegedly found spike protein to the Sanger sequencing because there may be full, correct integration in other cells that were not sourced, but again remember that the study did not look for these alleged cells, and so we are still left with the answer of “we don’t know”.
This correction should provide some context to #1 as it’s not an accurate rebuttal to the study since both can exist without contradiction one another- it may mean that the cells being sourced may not be the correct ones. Granted, it may also mean that integration isn’t happening at all. It requires that the authors provide clear evidence in support of their assumption.
This also still leaves the issue that the sequence is super messy, to the extent that it’s hard to argue that this is any validation of integration. Again, there are several issues with the sequence alignment that poses many questions.
Consider this a follow-up to yesterday’s post. Hopefully this will clear up some things that may have been confusing from yesterday’s post. I’ll blame any confusion portions of my article yesterday on me falling ill post-Christmas. What a gift that is!
Here is the post in question for those interested:
Do you know what you're looking at?

For those wanting a quick tl;dr a study suggesting genomic integration of the mRNA vaccines doesn’t actually show that, but in fact likely doesn’t show anything related to the vaccines. I’m partially…
1. Findings that contradict one another
Yesterday I raised questions regarding the mass spec results. Here, the authors took serum from patients and treated them to trypsin to digest the proteins. The authors allege that the mass spec results could find distinct patterns between virus spike and vaccine spike, looking in particular at peaks for 830.3 and 979.4 m/z in particular.
I don’t know why these peaks were used in particular, but let’s assume that these peaks were the result of previous studies looking at vaccine spike and their fragmentation patterns when treated with trypsin.
If true, then these findings may suggest constantly circulating spike, but the authors don’t provide us any reasons to believe that we can reach this same conclusion as them.
What’s made worse is that their sequencing results would actually contradict these findings.
Now, let it be clear that these results are not what the researchers are making them out to be- I don’t trust that the alignment is accurate and I explained why in the prior post (and will further elaborate in this post) but again let’s assume that the amplicon sequence is accurate:

Even if we are to trust the amplicon sequence (the bottom sequence in (B)), then we can clearly see there are parts in which the bases from the amplicon don’t match the bases from the Pfizer sequence.
This may seem like nothing important, but remember that DNA → mRNA → Proteins. That is, any places where there are mismatches in the amplicon sequence relative to the reference Pfizer sequence, we have to speculate that this may code for a different amino acid, and if so then the molecular weight of the spike protein would be different. This would be even more so if the base changes would remove any lysine or arginine residues in the protein since those are trypsin digestion sites.
All this to say that we can’t accept both experiments as being accurate because both would contradict one another. If the mass spec results are reliable then that would infer that the integrated sequence is a 100% to the Pfizer sequence, otherwise you would be making a different protein with different fragmentation patterns. But if the Sanger sequence is accurate then it tells us that we must accept the base mismatches between the Pfizer sequence and the amplicon sequence, which again would suggest that the mass spec results may not be trusted.
Both results cannot be accepted because both results would contradict one another.
Of course, a rebuttal to this may be that the mass spec results are qualitative. The authors appear to be looking at percentage of each m/z peak, but again without further clarification we as readers can’t speculate on how to interpret the data. It should be made clear by the researchers.
2. Those are funny looking bases
Now, there’s one thing I didn’t clarify in yesterday’s both that’s worth considering as a critique for the findings.
Aside from the typical G,A,T, and C bases one would see in sequencing you may have noticed a few other letters such as M, Y, V, S, and R (among a few), as boxed below:

These additional letter notations are referred to as “degenerate bases”, and usually refers to areas where several different bases may be “called” rather than one distinct base.
The nomenclature used by IUPAC is as follows (from Wikipedia):
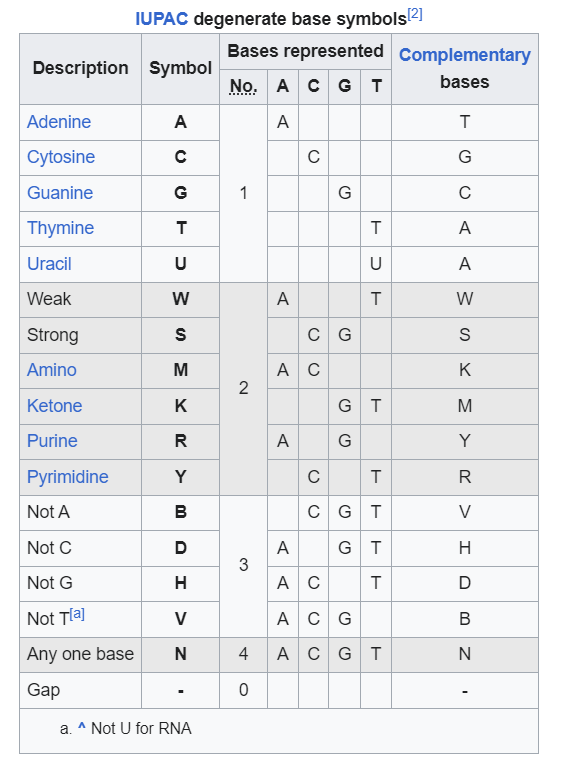
Now, in some of the articles I’ve seen they’ve commented that the two-base degenerate calls are a sign of messy sequencing product. However, there’s also a very clear explanation for that sequence as well, and that answer lies within our genomes.
That is, as a species we are diploid- we carry two sets of chromosomes rather than one or 3 as in some plant species. Because we are diploid we can exhibit heterozygosity, where a gene on one chromosome may code for a different genotype/phenotype as a similar gene on the other pair of the chromosome, usually stemming from different mutations within said gene.
These base differences in genes are called polymorphic regions, and can be elucidated by sequencing.
In this case, it’s not likely that those degenerate base calls are due to messiness, but are likely a sign that the sequence that was amplified- whatever it is- is polymorphic. Or put another way, the person who this sequence is taken from is likely heterozygous for whatever this sequence is.
For example, HLA, some of the most polymorphic genes we have, tend to show a lot of heterozygosity within the population. An example chromatogram, albeit a bit old, can be seen below where a “Y” and “K” are shown to indicate someone who is heterozygous for HLA-C alleles1:
. Heterozygous positions are denoted by letters other than A, C, G, or T; a Y at 538 represents the occurrence of a “C” and “T”, with the “T” contributing to the new tryptophan at amino acid 156 in Cw*16041.")
Of course, poor sequencing should be considered but for anyone who has done sequencing of human genes polymorphism are a given for these samples.
This actually raises even more critiques towards the idea that this is evidence of integration, because again remember that integration would require that an entire sequence (in this case over a 4,000 bp insertion) be integrated into the person’s genome. This is where the nebulous statement of integration ends up biting vaccine skeptics in the rear end, because without any clear context it becomes easy to suggest that anything can be an example of integration.
To me, what this sequence appears to suggest is not integration but likely that some polymorphic sequence was amplified.
Again, the authors could add further clarification for their sequence results, but the fact that heterozygosity is shown suggests that this isn’t a vaccine sequence that may have been amplified. Keep in mind that the primers the authors used appear to be nonspecific, and so it’s likely other genes may have been selected for and them amplified. This is why the lack of using additional primers for nested PCR is a problem- it removes much-needed filtering of off-products.
3. You shouldn’t be able to see this sequence.
As mentioned yesterday, further evidence that this alignment is incorrect is the fact that the allegedly homologous sequences like outside the amplified region. Remember that the “AGCAATT” sequence is found upstream of the amplified region, and therefore should not have been amplified through PCR. Put another way, there’s no way the authors would be able to see this sequence because it shouldn’t appear within their amplicon. This adds further evidence that they may not have amplified the end sequence of the vaccine as they assumed, and again likely amplified some native sequence within the patient.
It’s rather surprising that not many people who have reported on this paper have caught this discrepancy. The authors are expecting us to looking at an alignment that should not exist- the “AGCAATT” sequence should not exist because it’s not part of the region bound by the primers used!
And to add further evidence of this impossibility note the numbers above the sequences:
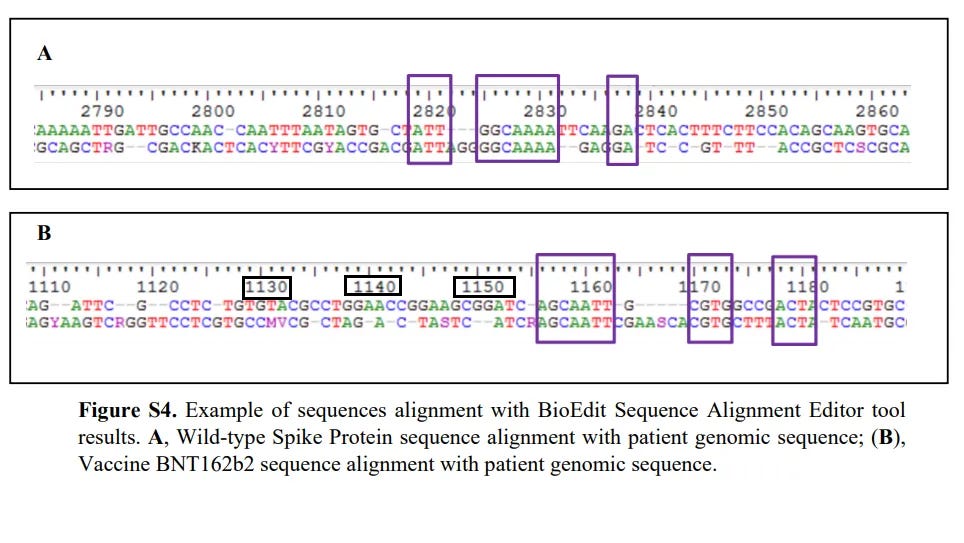
These numbers tell you the position of the bases being looked at. This adds further evidence that, for some reason, the authors aligned their amplicon with the beginning of the Pfizer vaccine sequence. We can verify this by looking at the sequence from github.
Note that every row is around 90 bp each. If we use the “AGCAATT” sequence as a reference sequence to look for we can see there is around 12 rows of bases, or around 1080 bases until you reach the correct row for the “AGCAATT” sequence:

Again, this adds further emphasize that the authors aligned their amplicon sequence not with the proper region of the Pfizer vaccine (i.e. the end) but rather aligned their amplicon with the beginning sequence of the vaccine. Remember- this shouldn’t be possible because this portion was not amplified by PCR!
I’m not sure why this may have happened. One possibility could be that the authors told the software to align wherever possible and they didn’t catch that the alignment was somewhere it shouldn’t be. There are other reasons that may be more cynical, but I won’t delve into that.
All this does is suggest that the authors didn’t find a sequence that they allege they did, and all the work around it obfuscates the fact that these are not matching sequences and is a clear indication that they did not confirm vaccine integration.
This further emphasizes that this study doesn’t prove much of anything, and yet I’ve seen several articles doubling down on these findings. Why is it that confirmation bias overlooks bad science? Shouldn’t it be clear that there’s something fishy with this study, so why is it that vaccine skeptics are still holding it up as a bombshell study?
So for a tl;dr:
The mass spec and Sanger sequence results are self-contradictory. Both can’t be simultaneously correct.
It’s very likely that the author’s did not amplify the Pfizer sequence, mostly because there lacks clear homology between the sequences. The heterozygous nature of the patient’s amplicon adds further suggestion that some native sequence was amplified.
The author’s own alignments shouldn’t be possible because their alignments fall outside of the region that they allegedly amplified. Several bits of evidence suggest that the alignment is incorrect including where the “AGCAATT” sequence is found and the base numbers which suggest once again that the alignment was done upstream of the amplified region. All of this warrants serious scrutiny of this study.
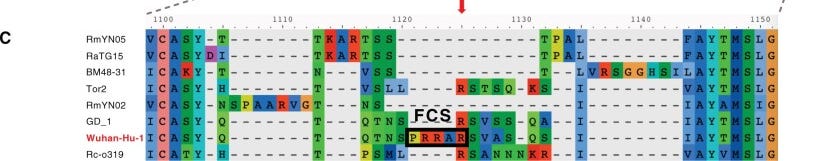
As Brian Mowrey mentioned in the comments yesterday you shouldn’t have gaps in the reference sequence. After all, you are referring your sequence to that original reference sequence. One area you may notice gaps is alignment of SARS-COV2’s spike with the spike of other coronaviruses due to the existence of the furin cleavage site, where in this case a gap for other coronavirus sequences are used to keep the rest of the sequence in alignment. An example can be seen below2, although note that this example is looking at amino acid sequence and not nucleotide sequence (the concept is the same). Note that this is intended to infer insertions/deletions of bases, which shouldn’t be the case for a reference sequence. This adds further scrutiny as to why these gaps in the reference sequence even exist.
If you enjoyed this post and other works please consider supporting me through a paid Substack subscription or through my Ko-fi. Any bit helps, and it encourages independent creators and journalists such as myself to provide work outside of the mainstream narrative.

Seán Turner, Mary E. Ellexson, Heather D. Hickman, David A. Sidebottom, Marcelo Fernández-Viña, Dennis L. Confer, William H. Hildebrand; Sequence-Based Typing Provides a New Look at HLA-C Diversity1. J Immunol 1 August 1998; 161 (3): 1406–1413. https://doi.org/10.4049/jimmunol.161.3.1406
Chan, Y. A., & Zhan, S. H. (2022). The Emergence of the Spike Furin Cleavage Site in SARS-CoV-2. Molecular biology and evolution, 39(1), msab327. https://doi.org/10.1093/molbev/msab327
More from Modern Discontent on that not-ready-for-primetime research supposedly showing integration of COVID jab material into human cellular DNA obtained from blood samples.
(I initially had seen insufficient procedural explanation, and insufficient alignment to make any claims, and therefore disregarded the conclusion of DNA integration. And this was without detailed knowledge of the analytical techniques currently used in such research.)
Modern Discontent tells you more about the analytical alternatives.
Should people keep looking for this DNA integration? Yes, because it's very possible, with the augmented delivery system, to get the bunch of DNA adulterants existing in the jabs into the cell and nucleus. (Never forget that the idea of RNA jabs was proposed as the "safer" alternative to circumvent genetic integration.)
We need independent analysts such as Modern Discontent to sift through ongoing research to discern which conclusions are well based in the data. Therefore, consider the power of the $ubstack $ubscription$ for keeping a watchful eye on the world of medical research and everything else.