


Do you know what you're looking at?
And why you can't always trust a researcher's word.

For those wanting a quick tl;dr a study suggesting genomic integration of the mRNA vaccines doesn’t actually show that, but in fact likely doesn’t show anything related to the vaccines. I’m partially inclined to believe that this study was an erroneous fishing expedition with some questionable results to infer integration is occurring.
Please if readers have any questions comment below as I understand if this article is rather confusing.
Correction 12.27.23: As Brian Mowrey mentioned in the comments note that the reference to the cells in this study are “leukocytes”. This is likely due to the cells that are being sourced, as blood samples were taken from patients. Note that my explanation is simplistic as it doesn’t take into account that the results are specific to leukocytes, as in we can’t infer what is happening in other cells. It also has to take into account whether the sample leukocytes are representative of all of the patients’ leukocytes as well. Because of these factors it isn’t correct to try and compare the allegedly found spike protein to the Sanger sequencing because there may be full, correct integration in other cells that were not sourced, but again remember that the study did not look for these alleged cells, and so we are still left with the answer of “we don’t know”.
This still leaves the issue that the sequence is super messy, to the extent that it’s hard to argue that this is any validation of integration. Again, there are several issues with the sequence alignment that poses many questions.
Since the start of this Substack it was my intent to examine many manners of COVID vaccines, COVID, and other topics related to health in science given that I have some background in the field.
However, while going down this rabbit hole what became extremely apparent was that even having a background in science isn’t enough to understand the vast wealth of knowledge and information available. It made me realize how ignorant and unprepared I was even carrying a degree in biochemistry and having worked in a lab for several years.
So part of this Substack wasn’t intended just to disseminate information to readers, but also served as a way to learn more about available science, and in particular become more adept at parsing studies and seeing whether studies are of merit, of some studies utilized flawed methodology, or if researchers may just erroneously make conclusions that are not supported by their own research.
It became part of my intent to educate readers so that they can parse information themselves- know how to break down what they are reading rather than being told what to think of a study. But again, such work requires plenty of time and effort, especially if it requires one to gain a ton of background knowledge in order to better break down a study.
Unfortunately, over the past few years I began to question how may vaccine skeptical people are engaging with evidence in the same manner- scrutinizing bad data and understanding what it is they are looking at when presented with a figure. Rather, it appears that confirmation bias can plague even vaccine skeptics; an issue that many groups may succumb to regardless of their stance.
If something appears to suggest some issue regarding the vaccines it may be picked up and used as a talking point even if the evidence isn’t there to support such a conclusion.
This brings us to the current predicament with this study1:

A lot of people have covered this study recently, using it as bombshell evidence that mRNA vaccine integration is occurring. Again, the question we need to wrestle with is not confirming the study for the sake of our own biases, but to see if the study is actually of merit.
As a foreword for readers, this study has plenty of issues, and honestly raises serious questions as to what exactly the authors were doing.
To start, the study looked at 81 people who are alleged to be Long COVID patients to determine where their Long COVID symptoms may be stemming from- either virus or spike derived or vaccine spike derived.
This is, right from the start, a problem as there is no explanation for what criteria were used to suggest that these patients were Long COVID patients. Because this study is looking at both vaccine and virus spike it’s also important to differentiate and categorize patients based on time of symptom onset and time from infection/vaccination, which again is not described.
Note that for the 70 patients where clinical data was known all 70 were previously infected whereas 51 (~73%) of patients were vaccinated:

This creates a problem because there’s no clear differentiation between vaccine or virus spike- it relies on the authors to look for something and to make an assumption about what they have found, essentially leading to a fishing expedition.
To that, the authors utilized mass spectrometry to look for fragments of spike protein in patient blood by first utilizing the protease trypsin in order to digest contents of the serum:
Mass spectrometry analysis was performed on the serum samples obtained from the recruited long-COVID syndrome patients, with the aim of detecting and quantifying spike protein fragments present in the samples. To achieve this, trypsin digestion was employed, generating specific tryptic fragments for each spike protein variant. The distinct tryptic fragments identified in the samples allowed for discrimination between the vaccine spike protein and the viral spike protein.
Trypsin is a protease that digests the carboxylate side of Arginine/Lysine residues.
Note that, protein-wise, the only difference between virus spike and vaccine spike is the swapping of two amino acid residues with proline residues, with the assumption that the proline residues would lock the spike into one conformation.
In regards to the vaccine one lysine residue was swapped for a proline residue, which would suggest that peptide fragments derived from vaccine spike would be inherently longer, or at least would contain one peptide fragment that is longer than virus spike.
Now, the actual length of these peptide fragments are what’s important, and to that the authors don’t provide any explanation for why they looked at these two peptide fragments in particular.
In this case, the authors looked at peptide fragments with a mass/charge ratio (m/z) around 830.3 and 979.4.
In short, mass spectrometry is an analytical method in which a compound gains/loses electrons in some manner. This causes fragmentation patterns of said compound, but it also leads to the introduction of different charges into the fragments. A detector then detects for these charged fragments and elucidates their size. It’s important to note that mass spectrometry is dependent upon the charged nature of the fragments. That is, if a fragment has a molecular mass of 500 but carries two charges it may appear on a mass spectrum as 250 (500 divided by a charge of 2).
All this to say that without any additional context we can’t know for sure what we are looking at when it comes to these mass spec results, or what peptide fragments the 830.3 and 979.4 are referring to in particular. Initially, I assumed that the 830 referred to virus spike protein whereas the 979 was derived from vaccine spike, but now I am not sure, and again the authors don’t provide any explanation for why they looked at these peaks in particular.
And even here the results are not very clear. When looking at the trypsin-treated serum of the 81 participants the researchers allege that they found fragments relating to virus spike in two patients and vaccine spike in one patient:
Out of the 81 long-COVID patients analyzed, fragments of the vaccine spike protein were found in 2 patients, while fragments of the viral spike protein were found in 1 patient (Table II).
Note that the actual Tables don’t tell us anything aside from descriptions of the results, because what sort of signal should we actually expect to see? It’s important that researchers explain what exactly is going on here, and yet this is all the information that is provided.
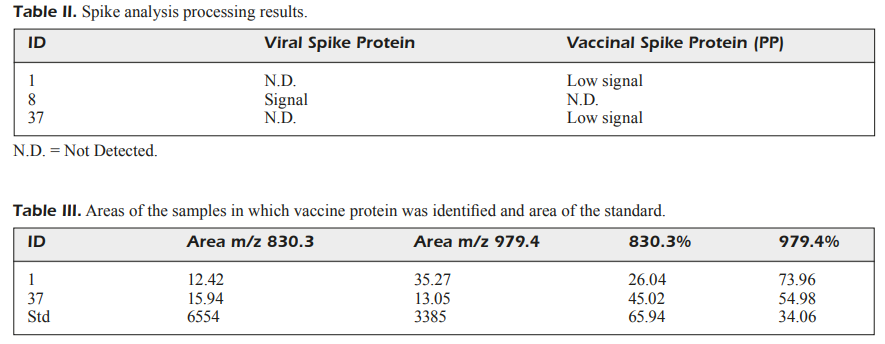
For all we know the results here may not pertain to any spike in particular. It could be any peptide fragment that the authors looked into. Again, it’s important that the authors explain why they chose these two peaks to examine.
So on its own this bit of information doesn’t help explain much because we can’t really tie the results to anything aside from having to trust that the researchers found evidence of continuously circulating spike (both virus and vaccine) even though this is “evident” in only 3 of the patients.
But that’s the least of the issues with this study. What’s rather concerning is the other experiment that the researchers conducted, and it’s here where a lot of problems arise.
In addition to the mass spec experiment the researchers also examined patient DNA for sequences that may pertain to the spike from the vaccine.
In doing so the authors conducted PCR of DNA extracted from patients:
The samples were obtained following informed consent and ethical guidelines. Genomic DNA was extracted from the blood samples using a commercially available DNA extraction kit according to the manufacturer's instructions (Blood DNA kit E.N.Z.A., Omega Bio-tek, Inc., Doraville, GA, USA or Exgene Clinic SV mini, GeneAll Biotechnology, Seoul, South Korea).
Note here that the authors use the phrase “genomic DNA”, and it’s from this phrasing that the authors may confuse naïve readers. Note that DNA extraction kits are generally non-selective. That is, most are designed to isolate DNA from whatever is within the sample. Or put within this context, there’s no way to extract a person’s DNA selectively for certain chromosomes or genes- it’s inherent within the extraction method that you are going to get genomic DNA for whatever you are extracting. It seems that some readers have looked at the word “genomic” and immediately thought of integration, when for all intents and purposes the word genomic is a nothingburger that just tells of something that most DNA extraction kids are designed to do.
The authors then conducted PCR on the DNA using primers alleged to be specific to Pfizer’s vaccine spike protein, with their results appearing rather sporadic. Note that we don’t know which vaccine some patients got, and 1/4 of the patients with available medical information didn’t even report being vaccinated.
The description of these results are as follows:
PCR amplification using the specific primers targeting the BNT162b2 vaccine spike protein sequence resulted in nonspecific amplification in 38 out of the 80 samples (Figure S1). No amplification was observed in 42 samples. A band of approximately 440 base pairs (bp) was detected, which corresponded to the expected size of the fragment amplified by the BNT162b2 primers.
There’s a few things worth noting here. Primers are oligonucleotides that are included during PCR to serve as a template for the polymerase. If no primers are used the polymerase won’t know where to bind along the DNA strand. Therefore, primers are used and are critical to the “annealing” step of PCR as the primers bind to specific segments of the DNA and allow the polymerase a site to start elongating.

Primers are critical to PCR and should be very specific to whatever gene you are looking at. If primers are nonspecific they could bind to any portion of a DNA strand, causing off-site amplification. The fact that so many nonspecific amplicons were found in the results may suggest that the authors used poor primers, but that may be the least of our concern.
Although the authors note that they amplified all 81 (80?) samples their results only show results for 18 samples when they ran their samples on a gel using gel electrophoresis:

Note that gel electrophoresis is a qualitative analysis. In general, you may run a gel to see if you have any amplicon at all. Otherwise, if no bands appear something may have gone wrong during PCR. You should also expect to see bands around your expected amplicon size, but remember that these are qualitative.
Note that gels are run with a DNA ladder. A DNA ladder is a collection of DNA fragments of various known sizes which serve as a frame of reference for your own aplicons. It’s needed because you otherwise won’t know what to compare your band sizes to. Here, it appears the researchers used a 1 kb DNA ladder, which would include DNA fragments in multiples of 100 bp starting from a 100 bp fragment and going up to 1000 bp, or 1 kbp.
Because the amplicon from the vaccine spike (at least using the primers that the authors did) is expected to be around 440 you would check to see if your amplicons fall between the 500 bp and 400 bp reference bands from the ladder.
Strangely, given the results above we don’t actually see any amplicon/bands around the 500/400 bp mark, but rather between the 600-700 mark. We can infer this given that the labeled 500 bp and 400 bp ladders fall below where the actual amplicon bands are, so this doesn’t quite correspond to the 440 bp that the authors suggest that they detected.
This could be the nonspecific amplification that the authors were referring to, but it again raises some quality issues. It also doesn’t help that the description of the results detail how many samples show either no amplification or nonspecific amplification, and yet the number of samples showing the intended 440 bp amplicon is never provided.
In order to solve this problem the authors turned to nested PCR. Nested PCR can be considered a two-step PCR method, in which two different sets of primers are used for each PCR step. A researcher may conduct nested PCR when one set of primers is nonselective. With the use of two sets of primers a researcher can filter out off-products since it would be less likely for nonselective amplicons to share two sets of primer binding regions as the intended sequence. This should, hypothetically, lead to cleaner products:
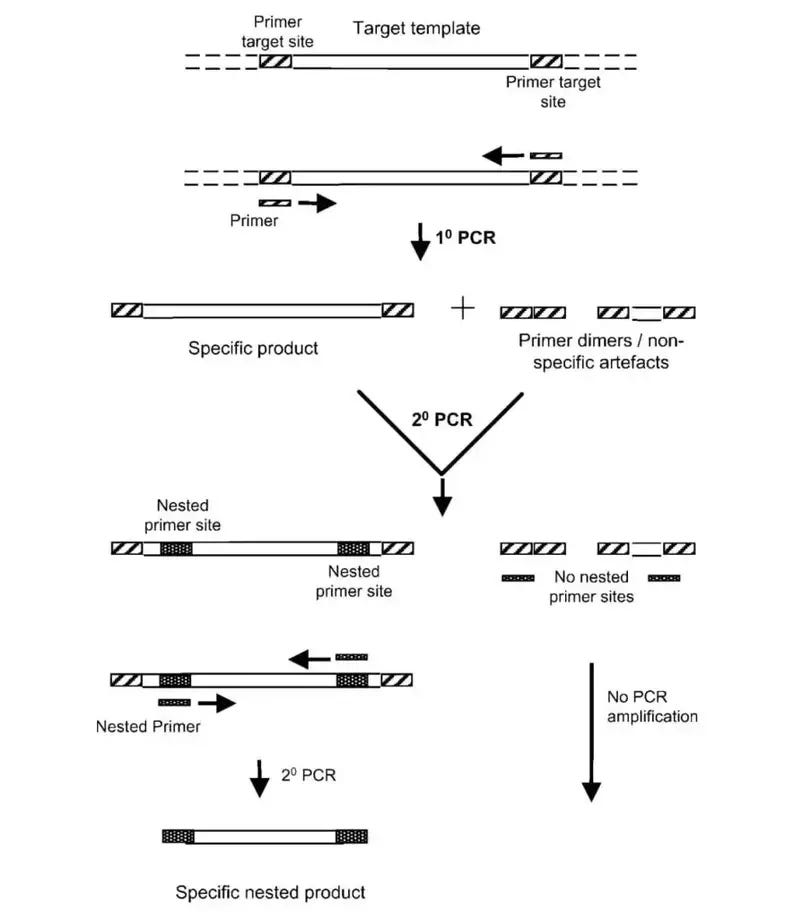
Note here that the significant aspect of nested PCR is the fact that two sets of primers are used.
Although the researchers conducted nested PCR they used the same set of primers twice (emphasis mine):
Due to the presence of nonspecific amplification in the initial PCR analysis, two rounds of Nested PCR were performed. The same primers and PCR conditions were used for both rounds. DNA extracted from the gel bands was purified using a gel/PCR extraction kit.
This is dangerous because if your primers are nonspecific to begin with nested PCR would only serve to continuously amplify these nonspecific amplicons because you wouldn’t provide a way to better select for the intended sequence.
This is made even worse given the description of how the authors included the samples that they did for the nested PCR step (emphasis mine):
To obtain specific and reliable results, two rounds of Nested PCR were performed on 12 selected samples that showed quality bands after the initial PCR (Figure S2). The second round of Nested PCR successfully amplified a single band of approximately 440 bp, closely matching the expected size (Figure S3).
What this appears to suggest is that the researchers didn’t choose ALL samples to conduct nested PCR on, but instead they seem to have selected for samples that show the 440 bp fragment size that they were looking for.
Even then, the two rounds of nested PCR still show a bit of nonspecific amplification, with a strange appearance of two bands outside of the 500-400 bp range in Figure S2:

So there’s already quite a few problems going on here. I’ll look closer into the primer sequence further on, but for now we’ll point to their Sanger Sequencing results, and it’s here where I have serious concerns for the author’s results.
As noted above gel electrophoresis is a rather qualitative approach in examining your amplicon. It only tells you the approximate range for your amplicon using the DNA ladder as a reference, but it can’t tell you what the exact size of your amplicon is, nor can it tell you the sequence of that amplicon.
One way of elucidating the sequence is to use a methodology called Sanger Sequencing. You can consider Sager Sequencing as gel electrophoresis with far better fragment separation and sequence analysis.
In short, Sanger Sequencing is a method in which you take your PCR product and undergo PCR one more time. However, this second round of PCR is rather different. Rather than just add in the typical reagents, an additional set of reagents utilizing truncated dNTPs (deoxynucleotide triphosphates, or the bases that make the building block of DNA) are included.
These truncated dNTPs contain modified 3’ regions and also contain a fluorophore (color-containing molecule) distinct for each modified base. During this second round of PCR some of these modified dNTPs may be added to the growing strand of DNA. However, due to their modifications they can’t continue their elongation, and essentially their inclusion into the DNA fragment results in elongation stopping abruptly.
The end result is that you have a stew of differently sized DNA fragments, and it’s here where a specific instrument known as a sequencer is used. A sequencer is a far more fine-tuned gel, in which the capillaries of the instrument are able to separate these many DNA fragments not by approximate size but by single base differences. That is, a sequencer can discern a 501 bp fragment from a 502 bp fragment, and can essentially separate all of these DNA fragments into their appropriate sizes.
Afterwards, the instrument is able to discern the last base in the sequence fragment due to the fluorophore.
For a clearer description ThermoFischer details Sanger sequencing in the following manner:
In order to determine the sequence, Sanger sequencing makes use of chemical analogs of the four nucleotides in DNA. These analogs, called dideoxyribonucleotides (ddNTPs), are missing the 3´ hydroxyl group that is required for 5’ to 3’ extension of a DNA polynucleotide chain. By mixing ddNTPs that have been labeled with a different color for each base, unlabeled dNTPs, and template DNA in a polymerase-driven reaction, strands of each possible length are produced when the ddNTPs are randomly incorporated and terminate the chain. The extension products are then separated by electrophoresis, resolved to single-nucleotide differences in size. The chain-terminated fragments are detected by their fluorescent labels, with each color identifying one of the terminating ddNTPs. The sequence of the template DNA strand can thus be derived by analysis (Figure 1).
The end result is that a chromatogram is constructed noting various colors at various base sizes, with software being used to discern what bases in particular are fluorescing:

So by utilizing Sanger sequencing a researcher can determine the actual sequence of their amplicon, and unfortunately this leads to probably the most egregious problem with this study.
First, let’s turn to the primers that the authors use, because this will tell us what sequence we should expect for the amplicon (emphasis mine):
PCR (Polymerase Chain Reaction) was performed using specific primers designed to target the spike protein sequence derived from the BNT162b2 vaccine (BioNTech/Pfizer mRNA Vaccine) (1). The forward primer (CGAGGTGGCCAAGAATCTGA) and reverse primer (TCTGGAACTAGCAGAGGTGG) were used.
The forward primer allows us to find the sequence within the Pfizer sequence. Here, we’ll use the sequence provided on github since that’s the sequence the authors use. You can find that sequence shaded below:

Note that this primer is intended to bind near the end of the spike sequence. This seems to be due to the fact that the authors are using a similar forward primer that was used by Aldén, et al.2 which is intended to target the end of the vaccine sequence:

The reverse primer is a bit difficult. In this case, you need to swap all the bases for their complementary base and read the sequence backwards. This should give you the following sequence to search for:
CCACCTCTGCTAGTTCCAGA
Which can be found here within the 3’ UTR portion of the vaccine sequence:

Overall, what this tells us is that the amplicon found from the researchers should align with a sequence near the end of the sequence uploaded onto github.
And for further emphasis note that this is what the authors outline for their Sanger sequencing protocol:
The PCR products obtained from the Nested PCR were subjected to Sanger sequencing to determine the nucleotide sequence of the amplified fragment. Sequencing was performed in both the forward and reverse directions using fluorescently labeled dideoxynucleotides and a DNA sequencer. The obtained chromatograms were analyzed to confirm the presence of the BNT162b2 vaccine spike protein sequence in the genomic DNA of long-COVID syndome patients.
With on example sequence being shown below:

We’ll focus on (B), and note that the boxes point to areas of alleged homology between a sequence found in the vaccine vs the one from in one patient. Upon first viewing the boxed homology may seem scary, until you realize that there are several areas where there is no homology (i.e. the letters don’t match between one sequence and another).
This raises a lot of questions because if people are alleging vaccine integration then we should expect near 100% homology between these two sequences- the sequence shouldn’t vary greatly after integration, otherwise you wouldn’t be producing the same spike protein which the authors allege to have found with their mass spec results.
But upon further examination I wanted to look for where this one of these homologous sequences may be within the Pfizer vaccine sequence. In this case I wanted to see where the “AGCAATT” sequence appears. Again, remember that the amplicon should only reside near the end of the vaccine’s sequence.
But where does this homologous sequence appear? Strangely, rather early on in the vaccine sequence:

Weird, this shouldn’t be the case because the sequence should lie in between the two primers because that is what is being amplified. Readers can search for themselves, but I couldn’t find this sequence within the amplified region, and instead found it outside that region, which again shouldn’t be possible.
This raised serious red flags for me because how could the authors exactly find a sequence that they didn’t amplify? Remember that the authors themselves allege that they conducted Sanger sequencing using PCR product derived from their two primers, which again should only amplify the end of the vaccine spike sequence.
But the questions don’t end there. Afterwards I tried looking for the other homologous region “CGTG” to corroborate where this sequence is. Again, if we use the Pfizer reference that the author used we should find this sequence a few bases downstream from the “AGCAATT”.
But where exactly is this sequence? Not a few bases down, but in fact right next to the “AGCAATT” sequence (“AGCAATT” boxed in black and “CGTG” boxed in green):

This again, doesn’t make sense because the alignment shown above would suggest that these homologous sequences should be around 10 bases separated from one another- not right next to each other, so what exactly is going on?
This led me to look at the sequence they provide and elucidate what those dashes are within the sequence (boxed in red):

Now, there are several issues that may arise with Sanger sequencing which may lead to what are called “uncalled bases”. This can occur when your peak from your chromatogram is too off-center, in which case the software doesn’t know that a base is there and leaves it either blank or with the letter “N” for “not called”. However, if your sample is super noisy then the software may not be able to discern the proper call, in which case it may place several “N” calls rather than giving a clear answer, as is the case in the example below:

Initially I thought that these gaps were similar to these “N” calls in which case the author’s chromatogram may be more noisy than they expected. This would usually lead one to repeat the sequence, or to go in and make the calls themselves.
But given the base discrepancy between the homologous sequences from the author’s and from github this doesn’t appear to be the correct answer. That is, if a non-call was made for these bases we should expect a fairly decent size gap between the homologous sequences- the bases are there, but the software cannot make a call.
But this doesn’t appear to be the case because the homologous sequences are still there, but far closer together. This made me believe something else was going on.
To that, let’s remove the dashes and see what sequence we get. I’ll start from the “AGCAATT” sequence and move downstream/ to the right.
This gives us a sequence of:
AGCAATTGCGTGGCCGACTACTCCGTGC
And if we search the github sequence we get:

Yupp- if we remove the dashes we get the actual sequence found on github.
This tells us one thing- the dashes are not uncalled bases, but are gaps acting as spacers between the bases.
So there are not missing/uncalled bases, but for some reason instead the bases are spaced out to appear as if there are bases there.
What’s rather interesting is that the software used by the authors allows you to insert/remove gaps in sequences:
So why is this a problem?
It’s a problem for the main fact that this tell us that the Pfizer sequence and the alleged patient genome sequence are not in fact in-alignment.
Essentially, this tells us absolutely nothing because by all accounts these sequences are useless. They don’t in fact show homology. Unless, of course, you include spacers/gaps…
If we remove the gaps from the patient genome and the Pfizer sequence, then try to align them what do we get?:
AGCAATTGCGTGGCCGACTACTCCGTGC
AGCAATTCGAASCACGTGCTTTACTATCAAGTC
Note that I’m using the “AGCAATT” sequence as a frame of reference, not because I believe this is a homologous sequence. And by removing the gaps you actually remove the two downstream homologous sequences, which adds even further evidence that these sequences were likely never homologous to begin with. Even worse, the sequences used as a reference in the Pfizer sequence shouldn’t even be found in the patient because they didn’t even amplify the pertinent sequence.
What all this tells me is that the findings here are useless- there are too many contradictions, questionable sequences used, and questionable alignment methods. Note that I’m not suggesting that the authors have inserted gaps to create the appearance of alignment, but it nonetheless makes me very suspicious of what methods they actually used and how they achieved their results. With that, the degree of ambiguity with this study is enough to warrant serious criticisms.
So what did the authors find here? I’m not sure, but something tells me that it’s just as likely some native human sequence was amplified and led down this unnecessary rabbit hole. There’s no evidence that the mass spec and sequencing results are actually related to the vaccines, and the fact that this isn’t further explained should raise serious issues with this study.
I have plenty of other issues with this study, but I’ll leave them be for now. I’ll just make note that it’s seriously disheartening that such a study has been widely disseminate without so much as a serious degree of scrutiny. It’s telling how readily people have accepted these findings without question if they are actually possible.
If you enjoyed this post and other works please consider supporting me through a paid Substack subscription or through my Ko-fi. Any bit helps, and it encourages independent creators and journalists such as myself to provide work outside of the mainstream narrative.

Dhuli, K., Medori, M. C., Micheletti, C., Donato, K., Fioretti, F., Calzoni, A., Praderio, A., De Angelis, M. G., Arabia, G., Cristoni, S., Nodari, S., & Bertelli, M. (2023). Presence of viral spike protein and vaccinal spike protein in the blood serum of patients with long-COVID syndrome. European review for medical and pharmacological sciences, 27(6 Suppl), 13–19. https://doi.org/10.26355/eurrev_202312_34685
Aldén, M., Olofsson Falla, F., Yang, D., Barghouth, M., Luan, C., Rasmussen, M., & De Marinis, Y. (2022). Intracellular Reverse Transcription of Pfizer BioNTech COVID-19 mRNA Vaccine BNT162b2 In Vitro in Human Liver Cell Line. Current issues in molecular biology, 44(3), 1115–1126. https://doi.org/10.3390/cimb44030073
I appreciate your time spent on this. Until multiple researchers have dissected and reported on a study I take it all with a can of salt. It was not clear to me even at the beginning, just what these authors were trying to show. If they can't be concise and clear then it speaks of muddying the waters to me. I'm not sure about the "long covid" being an issue anyway - many viruses have long effects, we just never made a big deal of them.
Lot of essential analysis in that 'problem' of distinguishing between the injected and natural Spike...Thanks for that part!
I do believe that was the main purpose for that article, to say there is a natural Spike out there and it is the reason for 'long covid', another whitewash, not to mention the overall deception to continue calling covid shots 'vaccines', where in fact, they are not. That's the biggest deception of all, by the entire medical establishment.