


Adding some context to the new Japanese ADE study
Taking a nuanced perspective and showing that there's a lot here to address and uncover.

Edit: The caption for Supplementary Figure 4A incorrectly stated that the K-ML2 line was the cell line with higher expression of ACEII and TMPRSS2 when it should have stated that the Clone 35 cell line shows higher expression. The caption has ben edited with corrections in italics to reflect this error correction.
Edit 9/20/2022: I forgot to include a formal citation for the Walls, et. al. study mentioned near the end of the article. The citation has been included as Footnote 6.
This morning a few different Substacks have reported on a new Japanese study examining antibody-dependent enhancement (ADE) in vaccinated individuals as well as from previously deployed monoclonal antibody therapeutics.
Some noteworthy Substacks include Naked Emperor’s as well as Eugyppius (Naked Emperor appeared to have been ahead in the reporting). Take a look at the provided links for their perspective.
In any case, the search for concerning features of these vaccines has brought up ADE as one of the most probable aspects. It’s interesting that Fauci himself, in March of 2020, even commented that ADE is a viable concern for vaccines and something that needed to be addressed (timestamped below):
So what exactly did this Japanese study find and of what use are these findings for those who are vaccinated as well as those who many have received monoclonal antibody treatments?
What is Antibody-Dependent Enhancement?
In discussing ADE it’s worth explaining what ADE actually is.
One critical feature of antibodies that have been overlooked is the actual purpose of antibodies. Usually we have taken antibodies for granted as little Velcro-like units that float in our bodies until they adhere to a pathogen.
Depending on how the antibody binds that may stop the pathogen from entering into cells by acting as a barrier between the cell and the pathogen.
However, the antibody does not work alone but in facts work in signaling to other parts of the immune system in what’s called an effector function.
This is carried out by the end stalk of the antibody known as the crystallizable fragment (Fc region) portion. This region comes at the tail-end of the antibody, opposite that of the paratopic region (Fab region) which binds to the antigen of a foreign molecule or pathogen.
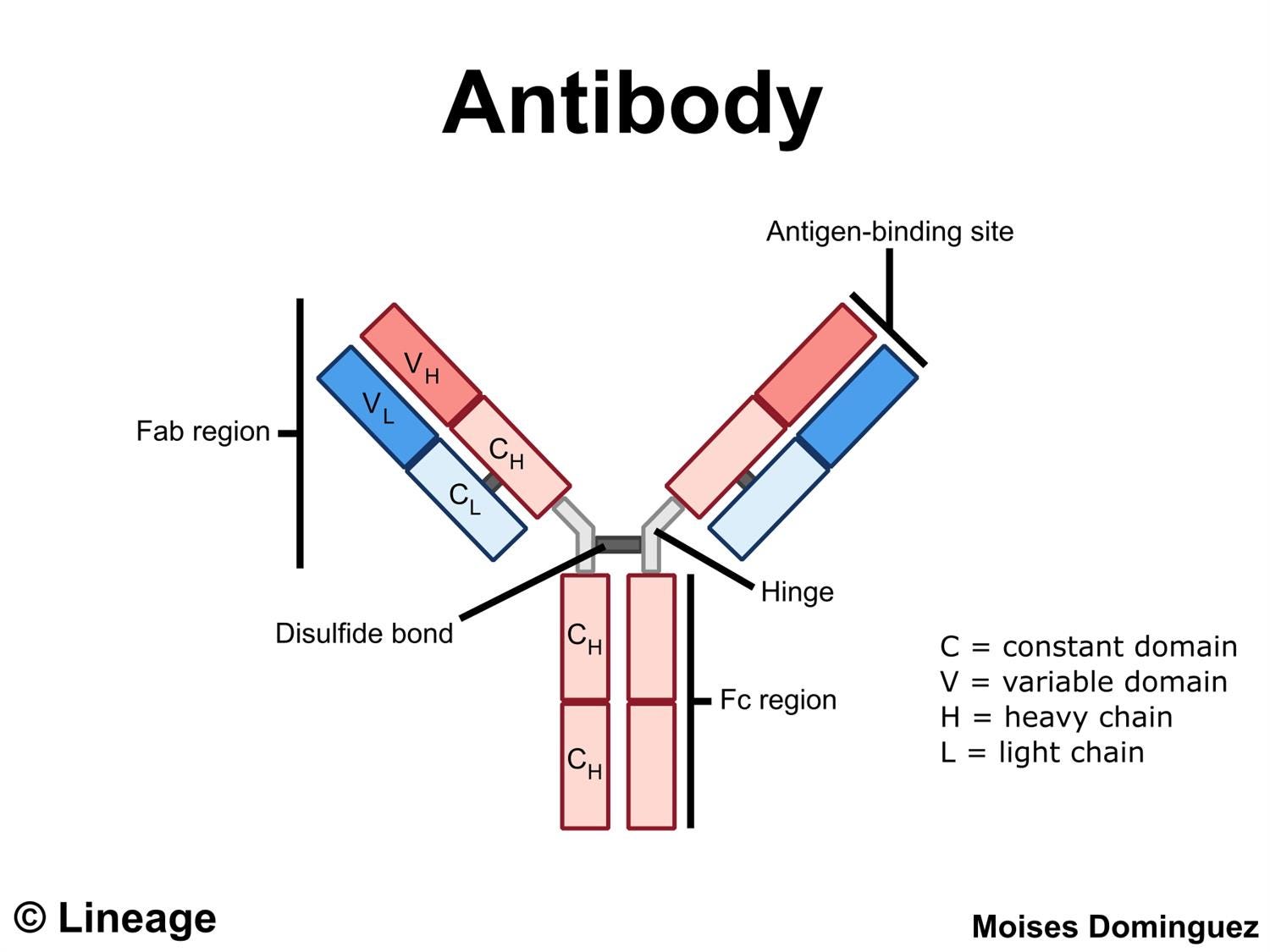
Now, when an antibody binds to a pathogen one of our immune cells, such as macrophages, monocytes, or other cytotoxic immune cells may bind to the Fc region and signal these immune cells to remove the attached pathogen. This binding occurs due to the presence of Fc receptors on the surface of these immune cells.
Such a response is considered to be an Fc-dependent or Fc-mediated immune response. It should be noted that other responses to antibodies may occur without binding to the Fc region (Fc-independent).
In most cases this response is well and good and leads to the removal of these pathogens.
However, in some cases it is the Fc-region which itself is culpable of increasing viral entry in a rather paradoxical sense.
Rather than hinder viral entry, the Fc-region of some antibodies, when bound to Fc receptors (FcR) on the surface of FcR-expressing cells may actually signal the cell to take in the virus rather than to eliminate it (Xu, et. al.1):
Fc receptor (FcR)-mediated ADE is the most common form of ADE, which was first discovered by Halstead (1977). At first, it was thought that the antigen antibody immune complex formed by virus and specific antibody combined with the host cell with the help of FcR on the cell surface, which was more conducive to the entry of virus, and increased the infection amount and infection rate of virus, finally led to the increase of infection and replication of virus. By phagocytosing immune complexes, the cells expressing FcR on the surface, such as monocytes, macrophages, dendritic cells, and certain granulocytes, can produce ADE. This kind of ADE is mainly mediated by IgG antibody, but IgM, IgE and IgA antibody have also shown the ability of ADE (Janoff, Wahl, Kelly, & Smith, 1995; Shi et al., 2018; Takada, Ebihara, Feldmann, Geisbert, & Kawaoka, 2007).
Below is a schematic diagram (Arvin, et. al.2) of some of these effector functions of antibodies, with ADE being boxed:

In many cases why this mechanism occurs has not be fully understood and tends to be muddied by the paradoxical immune response. On one hand, the antibodies that bind and eliminate a pathogen may effectively do so, but on the other hand at some point this may reverse and become detrimental. The exact reason why something that would be protective may be harmful makes research into ADE very nuanced and complex.
Things to Consider
Remember that in many cases the argument over ADE tends to be very, very broad. Here we’ve commented that an antibody needs to bind to the virus/pathogen first in order for the proceeding events of ADE to occur. That tells us that the antibody is critical in examining whether ADE is occurring, such that one antibody binds differently than another and affects which antibody may cause ADE in the first place.
At the same time, many different antibodies are all attempting to cling to an antigen/pathogen, so to the extent that ADE is occurring requires an examination of all the antibodies available and how they all may bind in concert to the antigen.
Keep this in mind as we look at the Japanese study.
The ADE Study
With this information in mind let’s examine the Japanese study circulating around.
The study in question is the very recently published article from Shimizu, et. al. 20223:

This study looked at ADE occurrence in two different experiments; one looked at monoclonal antibodies previously used for COVID and one looked at the sera of a very small pool of Moderna-vaccinated individuals.
What cell lines are used?
The cell lines used were myeloid cell lines, which are cells that differentiate into other immune cells such as macrophages and monocytes. However, here these cell lines were modified to express ACEII and TMPRSS2 (the serine protease that cleaves the spike into the S1 and S2 subunits) and labeled Clone 35 throughout the study:
Briefly, immortalized myeloid cell lines were established by the lentivirus-mediated transduction of cMYC, BMI-1, GM-CSF, and M-CSF into human iPS cell-derived myeloid cells. These cell lines were further induced to express ACE2 and TMPRSS2 using lentiviral vectors.
This is rather interesting. Remember that the remarks about ADE refer to Fc-expressed cells which usually focus on immune cells. To the extent that the virus alone can infect myeloid cells would depend on the expression of both ACEII as well as TMPRSS2 on the surface of these cells (note: myeloid cells do express ACEII receptors). By modifying these cells to express these receptors and enzymes this itself may induce greater infectivity of these cell lines.
Now, this is balanced by the use of these myeloid cells among control cell lines which would take into account this greater rate of infectivity, but it’s something worth remembering when examining this study and wondering how comparable this is to our own cells.
How was infectivity measured?
This study measured infectivity in a quantitative manner via the use of qRT-PCR.
Viral infection of SARS-COV2 into these Clone 35 cell lines should, in theory, lead to greater replication. Higher replication would infer greater viral load, and greater viral load would be measured by PCR:
Mylc cell lines (2 × 104/well) were cultured with SARS-CoV-2 virus in 96-well flat-bottomed plates. Viral concentrations in culture SNs after three days of culture were determined by qRT-PCR. Total RNA was extracted from culture SNs using the QIAamp viral RNA mini kit (QIAGEN) following the manufacturer’s instructions. […] The fold increases in viral quantity were calculated as follows: the fold increase = (virus concentration (copies/μL) of the experimental group with Ab)/(virus concentration (copies/μL) of the background control culture without Ab).
Which vaccinees sera was used?
This study focused solely on those who were vaccinated with Moderna’s vaccine. However, the pool was very small (6) but more importantly no patient information is provided on these individuals. No age, sex, prior infection history, or comorbidities were included either in the study or in the Supplementary Material, leaving us in the dark to possible reasons for variability.
This is a pretty big issue, because any variability in the data can’t be concluded to be of the vaccine alone, but without those other variables this makes it hard to fully extrapolate from this study.
*As a note, I will not go into too much detail in regards to the actual vaccinated individuals since other people have covered this section already.
Experiments on monoclonal antibody therapeutics
In this study researchers looked at the effects of monoclonal antibodies and their ability to induce ADE first. Two different monoclonals were observed; one combination therapy from Regeneron (REGN-COV) containing the antibodies Casirivimab and Imdevimab and GSK’s Sotrovimab (S230).
When examining ADE it’s important to note whether monoclonals have FcR binding capabilities (remember that it’s the antibody/antigen complex that interacts with the FcR on cell surfaces that’s important for ADE to occur). Some monoclonals may alter the Fc Region and prevent binding to receptors such as Evusheld’s prophylactic therapy that I discussed previously.
In this case the researchers found that all 3 antibodies were able to bind to the FcR of the Clone 35 cell lines as measured via immunostaining and flow cytometry, so we won’t discuss this part in particular.
Given that binding to FcR receptors by these monoclonals is possible, the researchers wanted to see if these monoclonals themselves would be capable of producing ADE.
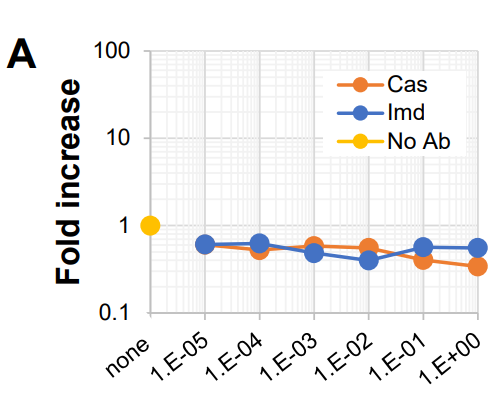
When examined at a high viral load both Casirivimab and Imdevimab didn’t appear to show ADE when cultured with Clone 35 cells at higher concentrations. However, at diluted levels there did appear to be an indication of ADE occurring:
Clone 35 cells were cultured with a constant dose of authentic SARS-CoV-2 virus (original strain: 4,000 copies/μL) in the presence or absence of a titrated amount of Cas or Imd Abs (Fig. 1A). Three days later, the amounts of SARS-CoV-2 RNA in the culture supernatants (SNs) were examined by quantitative PCR (qPCR). At high doses of mAbs, significant neutralizing activities were observed (for example, more than 90% neutralization at 100 ng/mL [1E-1], Fig. 1A). These neutralization efficiencies were consistent with those previously reported22, indicating that these mAbs in these experiments maintained their activities. However, at diluted concentrations (1 ng/mL final), both mAbs exhibited obvious ADE activity (Fig. 1A,B).
So remember that ADE is a measure of higher viral load measured 3 days after incubation as noted by qRT-PCR. Also, note that the metrics used in this study changes between the actual figure as well as the information in the text. For instance, above it mentions neutralization at 100ng/mL, yet the figure measures in micrograms (ug)/mL.
Remember that micrograms are a measure of 10^-6 while nanograms are a measure of 10^-9. The difference between nano and micro is a difference of 1,000 fold, and so 100 ng/mL is the same as .1 ug/mL which is the same as 1E^-1 on Figure 1A. Yes, very confusing!
And as stated above at a measure of 1 ng/mL (1E^-3) there appeared to be evidence of ADE occurring:

I’ll focus on this figure because I can’t quite make heads or tails as to what Fig 1C-E are intending to show aside from taking the fold measures from different studies and collecting them together so I’ll focus on 1A.
Now, this would appear rather concerning and show some sort of dose-dependent action. What action exactly is not described.
However, as a therapeutic one would wonder if this dose would be one achievable with the use of Regeneron. This would raise questions as to the practical findings of this in vitro study.
What’s strange is that the researchers note that this window of ADE appears to be very narrow and provides this graph within the Supplemental Material for Casirivimab specifically:

But what’s interesting is this comment made by the researchers:
The ADE observed for clone 35 cells was not observed for the parental cells, K-ML2 cells lacking the expression of ACE2 and TMPRSS2 (Supplemental Fig. 4A), and was blocked in the presence of an FcR-bindable and competitive Ab (4G2 mAb) (Supplemental Fig. 4B,C).
Above I mentioned that these Clone 35 cell lines were modified to overexpress ACEII and TMPRSS2 and that may significantly change the interpretation of results.
K-ML2 cells are the parent cells, meaning that they are the unmodified myeloid cells although they show rapid division. That means reduced expression of both ACEII and TMPRSS2 compared to the Clone 35 cell line.
Given this information, the researchers apparently didn’t find any evidence of ADE at various concentrations using the K-ML2 cell line:
And this makes sense, as another previous study by the same researchers looked at K-ML2 cell lines leading them to be surprised at the low level of viral infection when they used serial dilutions of SARS-COV2 (Shimizu, et. al. 20214):
In the present study, we used the Mylc line K-ML2 (Supplemental Fig. S1), because this line grows fast and it is easy to prepare a large number of cells. First, we examined whether K-ML2 cells can be infected with live SARS-CoV-2 (Fig. 1). K-ML2 cells were inoculated with serially-diluted SARS-CoV-2 and cultured for three days. Contrary to our expectation, no increment of SARS-CoV-2 in the culture supernatants (SNs) was detected by quantitative PCR (qPCR).
The researchers noted that, when ACEII expression of K-ML2 cell lines was examined expression was at a basal level (i.e. very low). This led researchers to try and upregulate the expression of ACEII using lentiviral vectors leading to the creation of the Clone 25 cell lines with an ACEII and TMPRSS2 expression level almost 100 times higher than the parent K-ML2 cell line:
Angiotensin-converting enzyme 2 (ACE2) is the binding receptor of SARS-CoV-2, and transmembrane protease serine 2 (TMPRSS2) processes the spike glycoprotein of SARS-CoV-2 for membrane fusion between the virus envelope and cellular membrane of target cells24. We next investigated whether K-ML2 cells express these molecules. qPCR experiments revealed that they express these molecules at almost basal levels (Supplemental Fig. S4). We therefore prepared ACE2- and TMPRSS2-expressing K-ML2 cells (K-ML2 (AT) cells, Supplemental Fig. S1) using a lentivirus expression system. The levels of expression of ACE2 and TMPRSS2 in K-ML2 (AT) cells were augmented more than 100 times (Supplemental Fig. S4) after lentivirus transduction.
So in some regard this sort of manipulation would actually not clue us in on ADE. If cells must be made to overexpress surface proteins can we really say that ADE is occurring?
On the contrary. Rather than argue that this low dose of monoclonals would lead to ADE the researchers, when comparing the K-ML2 cell line results with the Clone 35 results, actually suggest that these results are indicative of an ACEII-dependent method of ADE similar to their 2021 finding:
Taken together, these results demonstrate that Cas and Imd mAbs have the potential to cause ADE of infection at a particular Ab concentration, and that the ADE observed is dependent on ACE2 and FcR.
So what exactly does this mean?
It means that, on some molecular level, the interaction between the antibodies, the spike, and the ACEII receptor all need to act together in some unique way to increase the infective nature of the virus.
And that mechanism may actually be elucidated by the researchers looking at Sotrovimab.
The unique MOA of Sotrovimab
The researchers next looked at Sotrovimab and wanted to see if this monoclonal would show signs of ADE when used with the Clone 35 cell lines.
Interestingly, although Sotrovimab showed FcR binding it did not show any signs of ADE:
Sot mAb was able to bind to FcR as well as Cas mAb (Supplemental Fig. 5). However, in contrast to Cas mAb (Fig. 2A,C), Sot mAb exhibited no ADE activity at any Ab concentration examined and had neutralizing activity at a higher Ab concentration against both the original and Omicron strains (Fig. 2B,D).

So if Regeneron’s combination therapy of Casirivimab and Imdevimab may show ADE yet Sotrovimab doesn’t, what exactly is occurring here?
This would require us to understand exactly where these antibodies bind and how this feature is actually critical in examining ADE.
For both Cas and Imd both monoclonals bind to the RBD of the spike protein in nonoverlapping regions5:

This mechanism of action for Cas and Imd is not out of the ordinary- usually one would want antibodies to target the RBD of the spike in order to neutralize the virus.
However, at low doses this appears to have some effect on ADE and is related to ACEII in some fashion. It could be that the binding of these antibodies somehow, at very low doses, may infer a preferential conformational change in the spike that allows it to better bind to the ACEII receptor.
One hypothesis could be that both antibodies could bind to the FcR of a cell, and such binding may place the spike in the right position to then bind to the ACEII receptor and lead to viral entry, almost as if the cell is aiding in its own demise. A low concentration of the antibodies may indicate that several antibodies are needed to bind to the spike as one or two may actually provide enough space for the spike to bind to the receptor.
As of now there’s no information that I’m aware of in regards to this ADE mechanism of Cas/Imd, but with Sotrovimab it’s mechanism is one that likely explains the lack of ADE.
Sotrovimab is an interesting monoclonal. Unlike the others which were isolated from the convalescent plasma of previously infected individuals, or isolated from vaccinated then infected ACEII transgenic mice Sotrovimab actually came from an individual infected during the original 2003 SARS-COV outbreak.
The use of an antibody from SARS-COV rather than SARS-COV2 speaks of the conserved epitope on the spike protein such that Sotrovimab can work across these two viruses.
But what’s more interesting is that Sotrovimab doesn’t target the RBD, but a nearby loop. The targeting of this loop, strangely, causes a conformational change in the spike protein.
The spike exists in a prefusion/postfusion conformation. When the prefusion form attaches to ACEII it latches on and enters its postfusion conformation.
What Sotrovimab appears to do, by targeting this loop on the spike, is to induce the spike to take on a postfusion conformation.
One study by Walls, et. al.6 looked at the binding of S230 (Sotrovimab) to the spike of SARS-COV and compared this binding to SARS-COV and ACEII.
A few of the findings included:
Sotrovimab only bound to the spike in the open (prefusion) and intermediate conformations and not the closed (postfusion) conformation:
In contrast to LCA60 [another antibody the researchers were investigating], which could recognize all possible arrangements of the B domain, S230 only interacts with intermediate and open states, but not with the closed conformation. Since the interaction sites of both S230 and ACE2 are only accessible in the partially or fully open B domain conformations, binding of either of these two proteins to SARS-CoV S would sterically prevent sampling of the closed state (Figures 4A–4D).
The rest of the study is rather complex, but the findings were unprecedented given the fact that Sotrovimab’s binding almost mimicked the same effects as ACEII binding in that it caused the spike to enter into a closed, postfusion state:
Moreover, S230 (or ACE2) binding triggered the SARS-CoV S transition to the postfusion conformation. This finding is an unprecedented example of functional mimicry, whereby an antibody activates membrane fusion by recapitulating the action of the receptor. It remains to be investigated whether binding of S230 triggers virus-cell fusion when particles are bound to the surface of cells.
An antibody that mimics the effects of the receptor is striking since most antibodies are examined from the perspective of binding and blocking interactions. Yet here, Sotrovimab changes the conformation of the spike and renders it unusable.
And it’s likely this mechanism that differs between Cas/Imd. Even if Sotrovimab as FcR binding capabilities, it would essentially be presenting an inactive, closed off virus to the cell that isn’t capable of binding to the ACEII receptor. It may be this reason that Sotrovimab doesn’t appear to show any ADE as compared to the other antibodies that bind to the RBD.
I suppose Shimizu, et. al. 2022’s use of their Clone 35 cell lines would actually counter the comment raised by Walls, et. al. such that virus-cell fusion doesn’t appear to be triggered by additional ACEII and TMPRSS2 proteins. However, more research should help elucidate more evidence.
The sera of vaccinated individuals
The last thing worth examining is the vaccinated individuals. As this has been the focus of other Substacks I won’t dive too deep into this section.
What’s interesting is that the researchers, as noted by Eugyppius, appear to focus on one individual (HC2) and noticed a high level of ADE occurring within this individual. However, the data was highly varied among the other vaccinated individuals which doesn’t tell us much information.
More importantly, no comparison to naturally infected individuals was done which just adds more ambiguity.
Because of this it’s hard to argue whether ADE is indicative of vaccines or may be a consequence of just any immunological response to the spike.
One thing that many people seem to forget is that, for the most part, we are born with all of the antibodies that we will ever produce. That is, an infection does not lead us to create antibodies de novo, but rather that an infection causes our immune system to dive into our germinal centers and activate B-cells with the necessary antibodies.
So in some sense we can’t discount the fact that all of us have constructed our own unique array of antibodies, and this unique array may either help or harm us. It could be a fact that HC2 may have just been unluckily and produced those antibodies that cause ADE.
And that’s probably one of the most crucial takeaways from this study. Again, we must be careful in immediately making proclamations of vaccination and that natural infection would not lead to ADE.
Remember that all of the monoclonals in use, including the 3 from this study came from people who were previously infected (well, some may have come from mice I’ll concede that point). That means that all of us, even if naturally infected, may carry Casirivimab-like or Imdevimab-like antibodies, and that would mean that ADE may be a possibility for all of us.
Okay, that’s if we don’t take into account a dose, ACEII, FcR-dependent mechanism of ADE, or that these are just 3 of the many many different antibodies that our bodies are likely to produce, or that the actual expression of both ACEII and TMSPSSR2 may actually be low for myeloid cells.
As one antibody may infer ADE such as Casirivimab or Imdevimab, another may actually stop the virus from binding by changing the conformation a la Sotrovimab.
Remember to look at this information not within the context of how to target the vaccinated or the vaccine, but what this may mean for the own antibodies you produce. Remember to draw those relationships between monoclonal antibodies and our own.
Also, if anyone is curious some research has looked into mice who overexpress ACEII and found higher immune function with possible protective effects against cancer, Alzheimer's, and atherosclerosis7. This mice model may not be translatable to humans (especially in regards to inducing overexpression aside from genetic variances). Nonetheless, this may suggest that people with higher ACEII expression of myeloid cells may be protected from other diseases but may be more at risk of developing ADE.
It’s just another example of how the body and all of its processes are far more complex than we make them out to be, and something we should always keep in mind.
If you enjoyed this post and other works please consider supporting me through a paid Substack subscription or through my Ko-fi. Any bit helps, and it encourages independent creators and journalists outside the mainstream.

Xu, L., Ma, Z., Li, Y., Pang, Z., & Xiao, S. (2021). Antibody dependent enhancement: Unavoidable problems in vaccine development. Advances in immunology, 151, 99–133. https://doi.org/10.1016/bs.ai.2021.08.003
Arvin, A.M., Fink, K., Schmid, M.A. et al. A perspective on potential antibody-dependent enhancement of SARS-CoV-2. Nature 584, 353–363 (2020). https://doi.org/10.1038/s41586-020-2538-8
Shimizu, J., Sasaki, T., Koketsu, R. et al. Reevaluation of antibody-dependent enhancement of infection in anti-SARS-CoV-2 therapeutic antibodies and mRNA-vaccine antisera using FcR- and ACE2-positive cells. Sci Rep 12, 15612 (2022). https://doi.org/10.1038/s41598-022-19993-w
Shimizu, J., Sasaki, T., Yamanaka, A., Ichihara, Y., Koketsu, R., Samune, Y., Cruz, P., Sato, K., Tanga, N., Yoshimura, Y., Murakami, A., Yamada, M., Itoi, K., Nakayama, E. E., Miyazaki, K., & Shioda, T. (2021). The potential of COVID-19 patients' sera to cause antibody-dependent enhancement of infection and IL-6 production. Scientific reports, 11(1), 23713. https://doi.org/10.1038/s41598-021-03273-0
Hansen, J., Baum, A., Pascal, K. E., Russo, V., Giordano, S., Wloga, E., Fulton, B. O., Yan, Y., Koon, K., Patel, K., Chung, K. M., Hermann, A., Ullman, E., Cruz, J., Rafique, A., Huang, T., Fairhurst, J., Libertiny, C., Malbec, M., Lee, W. Y., … Kyratsous, C. A. (2020). Studies in humanized mice and convalescent humans yield a SARS-CoV-2 antibody cocktail. Science (New York, N.Y.), 369(6506), 1010–1014. https://doi.org/10.1126/science.abd0827
Walls, A. C., Xiong, X., Park, Y. J., Tortorici, M. A., Snijder, J., Quispe, J., Cameroni, E., Gopal, R., Dai, M., Lanzavecchia, A., Zambon, M., Rey, F. A., Corti, D., & Veesler, D. (2019). Unexpected Receptor Functional Mimicry Elucidates Activation of Coronavirus Fusion. Cell, 176(5), 1026–1039.e15. https://doi.org/10.1016/j.cell.2018.12.028
Veiras, L. C., Cao, D., Saito, S., Peng, Z., Bernstein, E. A., Shen, J., Koronyo-Hamaoui, M., Okwan-Duodu, D., Giani, J. F., Khan, Z., & Bernstein, K. E. (2020). Overexpression of ACE in Myeloid Cells Increases Immune Effectiveness and Leads to a New Way of Considering Inflammation in Acute and Chronic Diseases. Current hypertension reports, 22(1), 4. https://doi.org/10.1007/s11906-019-1008-x
"More importantly, no comparison to naturally infected individuals was done which just adds more ambiguity."
As well as naive control sera, to rule out nonspecific interactions. This is the same flaw as last year's "poised to acquire" paper (https://www.biorxiv.org/content/10.1101/2021.08.22.457114v1). If a study doesn't have a control, it isn't showing the effect of the intervention - just basic logic, and for some reason all of the ADE and half the imprinting papers flaunt it.
Wow, thank you for breaking this down in such detail. 👍🏽💕