


A failure in drug design amidst a pandemic- Remdesivir
Remdesivir was only administered intravenously, resulting in only hospitalized patients receiving the drug and possibly causing harm. Why was this the case?

In following the prior post this one is more for informative reasons. It’s well known that the drug Remdesivir was only administered intravenously, and given the series of events during the pandemic meant that Remdesivir was only provided to patients who were hospitalized with COVID, leading to possible complications, harms, and poor efficacy.
Now, I will leave the reasons for Remdesivir’s rushed EUA on limited data to others for now, and we’ll focus on taking a look at the intravenous use of Remdesivir because it highlights an important aspect in drug design.
Consider that most drugs are provided orally. Most people taking them may not think too deeply about this mode of taking a drug as it may seem like the obvious approach. But not all drugs are administered orally. Ozempic is given subcutaneously, nitroglycerine is generally provided sublingually, and some given rectally or through other openings in the body.
Part of this reason is based on where the infection/disease is and garnering quick access to this part of the body (eye drops to relieve pressure in the eyes for glaucoma, for instance), but part of this is based on how available the drug is for the body (called bioavailability), all of this relies on the pharmacokinetic properties of the drug- can it get to where it needs to in its proper form, or will most of it be metabolized or excreted before it has any effect on the body (first pass effect)?
Some of these topics were covered in previous articles for those who would like a deeper dive:
An Introduction to Pharmacology

I’ve always considered being a teacher and was considering going to graduate school to become a professor. Of course, life had a different idea in mind. Although I didn’t become a teacher I still lov…
But for now we’ll take a look at Remdesivir in particular and see how its development led to its intravenous-only use, and how this likely caused a host of problems amidst a pandemic.
A timeline of Remdesivir (as told by MD)
Note that this post won’t go too deep into the synthesis pathways of Remdesivir, but look more broadly at how its final design came to be.
The concept of utilizing nucleoside analogues has been around for years, and in part makes sense conceptually. If you can trick the virus or bacteria to take something up as a toxin then you can get it to kill itself, or halt its replication capacity.
There are a few problems with this model as our own cells rely on nucleosides as well, and these drugs can be nonselective in their targets. This is one of the issues with nucleoside analogues used for cancer treatment which indiscriminately target rapidly dividing cells such as hair, skin, nails, GI, etc.
Some examples of nucleoside analogues include the HIV drug Azidothymidine (AZT, or Zidovudine), which is an analogue of the native nucleoside Thymidine. The only alteration is the addition of an azide group on the C3’ position of the sugar ring1:

This is a bit of an overview, and we’ll focus more on Remdesivir down the line. Note a few liberties have been taken with how the actual process took place.
Now, let’s suppose that you are a fledging scientist (or group of scientists, or robots, or whatever really) working for a large pharmaceutical corporation with the hopes of ridding the world of diseases.
You first utilize computer models designed to figure out which modifications to native nucleosides will lead to effective candidates for targeting viruses (Eastman, et al.2):
As a starting point for discovery, a library of ∼1000 small molecules focused around nucleoside analogues was compiled, based on prior knowledge of effective antiviral compounds targeting RNA viruses. Nucleosides are poorly cell-permeable (and therefore can have a low hit rate in cell-based screens such as antiviral screens), so modified nucleosides such as monophosphate, ester, and phosphoramidate prodrugs composed a significant portion of the library. Such prodrugs are typically more permeable and metabolized to liberate the nucleoside or phosphorylated nucleoside within cells.40−42 While the data from the original full screen does not appear to have been disclosed, a 1′-CN modified adenosine C-nucleoside hit (GS-441524), along with a prodrug form of the monophosphate of GS-441524 (GS-5734, later renamed as remdesivir), was found to be highly potent.43
Here, you have two promising hits. One that appears to look like a nucleoside analogue of Adenosine3 which will be named GS-441524 as its code name. Why GS? Well, that is the company you work for (Gilead Sciences), so why not use that designation?
But there’s another hit for a monophosphate form you call GS-5734, later to be rebranded as Remdesivir.

Given these two options which one do you choose? Some drugs are administered in their nucleoside forms, although that comes with the risk of being recognized by host enzymes which may readily metabolize these drugs. There’s also a problem with bioavailability and uptake by cells (Mehellou, et al.4):
Nucleoside analogues are structurally different to natural nucleosides, making their phosphorylation by cellular or viral kinases often inefficient. (13,14) This ultimately limits the formation of the active triphosphate metabolites, often the nucleoside analogue active metabolite. (13) They also often demonstrate poor oral bioavailability due to their low intestinal permeability, as they are usually polar molecules that hinder their transport through the cell border via the paracellular route. (15)
In pondering this thought some more you come to recall an issue of the phosphorylation steps as well. At some point you heard that the first addition of a phosphate took the longest, also called the rate-limiting step [context added]:
This notion [using a monophosphate] was further supported by numerous studies that reported the poor phosphorylation of nucleoside analogues into their nucleoside monophosphates as the rate-limiting step in their in situ activation. (24−27)
The rate-limiting step in chemistry refers to the step that takes the longest among the other steps in a chemical reaction. It acts almost like a bottleneck to which other subsequent steps in a reaction get held up in.
To put it in laymen’s terms, let’s consider a recipe for cookies. We’ll go with this 72-hour from Jacques Torres published in The New York Times.
Let’s suppose we break down the recipe into 3 categories: prep, rest, and bake.
Prep and baking are quick and easy, let’s assume 10-15 minutes each (more near 10 on the baking side if I recall the recipe). Pretty quick, but then there’s that 72 hour period in the middle, where supposedly a lot of magic is taking place in this cookie dough that is worth all the waiting time.
This step is the rate-limiting step in this cookie making process. The prep and baking may be quick, but you’ll be caught up in the waiting period in between until the dough is ready. You can’t bake until the dough has sat the whole period.5 If you intend on making these cookies for a Friday potluck at work, you may want to get started on Sunday/Monday if you want to have the cookies ready by Friday.
This analogy is a bit of a stretch, but when it comes to the nucleoside or monophosphate form of Remdesivir the first phosphorylation is argued to be akin to the “72 hour resting period” for that particular cookie recipe. It’s assumed to be the step that takes the longest to occur. You can’t add additional phosphates until the first phosphate is added, and so if the first addition is the slowest then your antiviral will supposedly get bottlenecked at this step.
Remember that every nucleotide that gets incorporated into growing DNA/RNA strands are all in the triphosphate form, so you need to have all 3 phosphate groups added to the structure before it can be used by the virus.
Because of this rate-limiting step you decide to go with the monophosphate form, even if the evidence of the rate-limiting step in phosphorylation is considered controversial.
So now you have your model drug that is ready to be tested! At least, you would think so…
One problem with going with the monophosphate form of a drug is that the molecule is now not only more polar but also charged since the phosphate group is negative at physiological pH. This is a big issue, because charged molecules have a ton of difficulty passing through the lipid layer of cell membranes. There’s also the issue of the phosphate group being cleaved off as well, leading to the nucleoside metabolite form. Why go through all that trouble if the drug may be metabolized to the form you were trying to avoid in the first place?
However, the idea of using nucleoside analogue monophosphates as therapeutics faced two main challenges. First, these compounds would have poor in vivo stability due to dephosphorylation in the bloodstream, and second, their transport into cells will be inefficient because of the charged nature of the phosphorylated nucleoside.
So now there’s another issue- you have to get rid of the charges on the phosphate group. You can add on different structures, but if they can’t be cleaved off by enzymes then your drug ends up in a state of prodrug purgatory. You need to add something that can protect the drug, enter into the intended cells, and then come off to reveal the active metabolite.
So what groups do you add to your phosphate? This conundrum led to decades of research until a team of researchers led by Chris McGuigan at Cardiff University came up with the idea to add an amino acid ester as one of these charge maskers, as well as a phenyl group.
The result, called a McGuigan ProTide, has become the standard model for monophosphate masking.6 If anyone’s curious, the term “ProTide” is a portmanteau of “prodrug of a nucleotide”.7
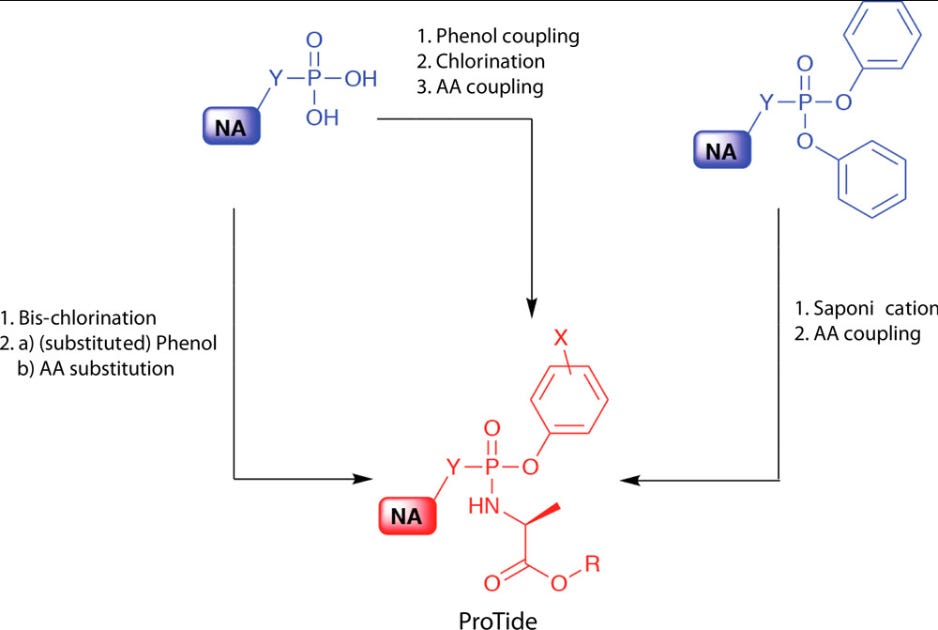
With the addition of the ProTide a nucleotide analogue can more readily pass into cells where the masking groups are enzymatically cleaved in order to reveal the monophosphate. This seems to occur using two enzymes: an esterase and a phosphoramidase:
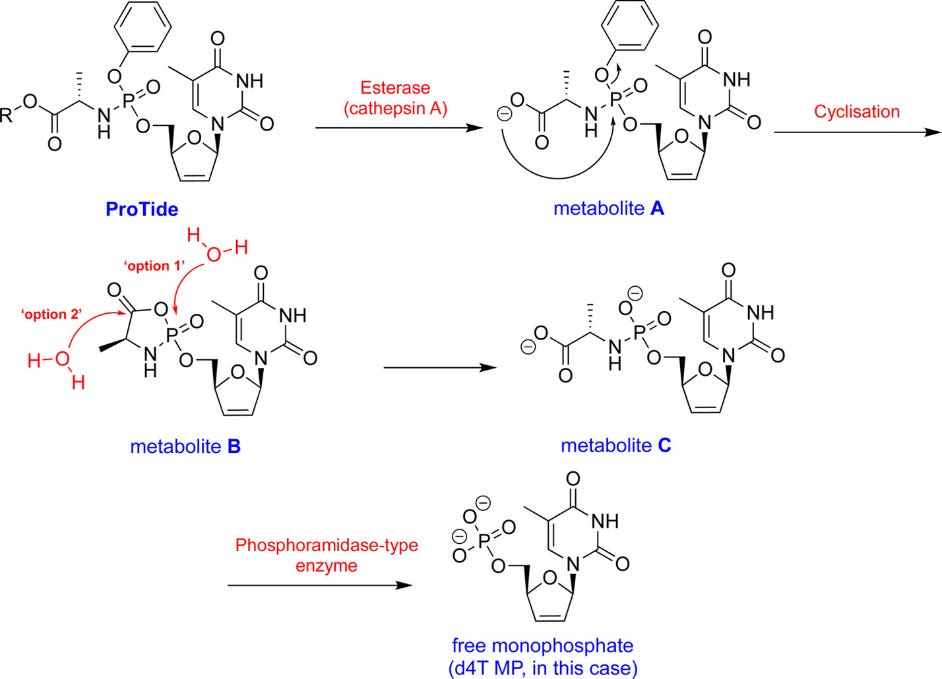
Alright, so you decide to go the ways of adding a ProTide, forming what would eventually be known as Remdesivir.
HOWEVER…
The addition of the ProTide group comes with its own issues. The removal of the charges don’t just make your drug more lipophilic, but now it makes it too lipophilic, to the point that it unfortunately has very poor bioavailability. Fatty/hydrophobic molecules tend to not want to pass into the bloodstream, and consequently are excreted readily from the body before eliciting their intended effects.
Now you have a problem- you have a drug but no real way to administer it without suffering bioavailability issues. You can come up with a lipid-encapsulated formulation, but that may be too difficult and the drug may still suffer poor bioavailability. One way is to get the drug directly into the veins, but there it will readily precipitate out of solution if not helped by some other substance.
The solution here is to turn to a carrier agent. A carrier agent acts almost like an emulsifier similar to egg yolks or mustard in a dressing. It has water-loving and water-hating properties, which both work to bring hydrophobic/lipophilic molecules into solution and marry together, again, similar to vinegar and oil coming together in a dressing if mustard is added.
In this case you turn to a carrier agent called sulfobutylether-β-cyclodextrin, or SBE for short.8
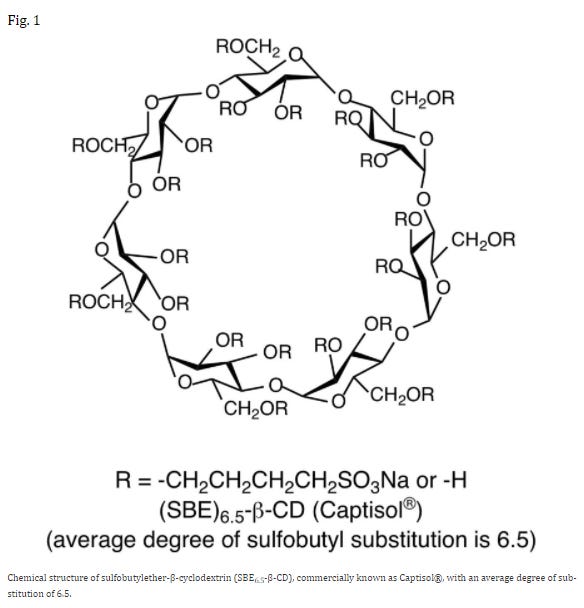
SBE has the properties of an emulsifier. It’s water-soluble due to the oxygens and charged sulfate groups while also containing hydrophobic regions that interact with Remdesivir. Paired together, SBE can bring Remdesivir into solution. This also means that SBE can stop Remdesivir from being caught up in serum proteins and lipids to some degree.
But because SBE needs to be dissolved in solution, the only way of providing Remdesivir is unfortunately through intravenous infusion alone.
Not suitable for a pandemic?
*A lot of information has been left out, including the testing of Remdesivir on various viruses. The focus here has been more about the process of why Remdesivir was designed in the way that it was.
All of this leads us into the conundrum of Remdesivir’s “compassionate use” for COVID. I suspect that because Remdesivir’s formulation and requirement of a carrier agent necessitated intravenous infusion, it had to be administered in a place that allowed for this process to occur. With everything shut down the only viable place seemed to be within the hospital, leading to the multitude of issues for an antiviral used so late in the disease progression.
With that being said, there are a few issues with this assumption. For instance monoclonal antibodies were provided to outpatients, so why wouldn’t Remdesivir?
Maybe it’s related to Remdesivir’s EUA approval acting almost like a Catch-22 situation. If it gets approval for use in a hospital it can’t then be carried over for use in outpatients, and so it gets stuck to only inpatient use where it was ineffective and more than likely harmful. Note that I am speculating here.
The need for repeat doses of Remdesivir also makes it more difficult to conduct with proper patient compliance. The only clinical trial for outpatient use of Remdesivir noted 3 consecutive days of intravenous infusion, which would likely be extremely difficult in the early days of COVID, and would be a burden for patients who are ill and would need transportation and time to visit the proper clinic.
Several investigations have looked at oral iterations of Remdesivir, but all appear to lack the monophosphate that Gilead seemed to want to include, with the most promising candidate seeming to be the nucleoside form GS-441524.
Again, note that this concept of the first phosphorylation being the rate-limiting step is actually controversial, with no clear evidence suggesting that to be the case. In fact, evidence suggests that the nucleoside form of Remdesivir appears to be more effective, as Remdesivir has trouble making its way into the lungs of patients.9 It also requires additional enzymatic steps that the nucleoside form would not have to deal with, ironically meaning that the addition of the ProTide actually led to possible bottlenecking.
In contrast, some models suggest that the nucleoside form is 100 times more water soluble than the ProTide form, which explains the need to use a carrier agent for Remdesivir and why the nucleoside form has been tested as an oral therapeutic.10
So maybe Gilead shot themselves in the foot by wanting to make a ProTide drug. As interest in the drug waned in the post-Ebola years and Remdesivir was shelved Gilead probably didn’t see a need to reinvent this drug and find oral formulations. When renewed interest in the drug came with SARS-COV2 Remdesivir could only be used in the way that it was formulated for.
I think there’s more to this story that can be discussed, and the ending portion here is more supposition. However, I hope it highlighted how such a minor change in a drug’s structure can have so many implications. Not only in administration of a drug, but in how and when a drug is used which can alter its safety and efficacy.
Substack is my main source of income and all support helps to support me in my daily life. If you enjoyed this post and other works please consider supporting me through a paid Substack subscription or through my Ko-fi. Any bit helps, and it encourages independent creators and journalists such as myself to provide work outside of the mainstream narrative.

Remember that the carbon, or C bonded to the oxygen and the nitrogenous base gets labeled as the first carbon, or C1’. The “prime” notation of ‘ is done to distinguish the organization of atoms relative to the nitrogenous base, which gets first dibs in numbering. Therefore, the sugar goes second and gets the carbons designated with a ‘.
Eastman, R. T., Roth, J. S., Brimacombe, K. R., Simeonov, A., Shen, M., Patnaik, S., & Hall, M. D. (2020). Remdesivir: A Review of Its Discovery and Development Leading to Emergency Use Authorization for Treatment of COVID-19. ACS central science, 6(5), 672–683. https://doi.org/10.1021/acscentsci.0c00489
The nitrogenous base of Remdesivir differs slightly from native Adenosine. Note that the Sugar-Base bond is a covalent C-C bond. Most Sugar-Base bonds are C-N bonds. This modification likely inhibits enzymatic cleavage of this bond as the C-C bond is stronger.
The ProTide Prodrug Technology: From the Concept to the Clinic Miniperspective
Youcef Mehellou, Hardeep S. Rattan, and Jan Balzarini
Journal of Medicinal Chemistry 2018 61 (6), 2211-2226
Technically you can make them prior to the 72 hour resting period, but from what I’ve heard the waiting makes a big difference.
During my schooling it was alleged that McGuigan sold the patent for this masking process, making either hundreds of millions or up to a billion dollars on this patent. I can’t recall the actual figures, but it makes sense why so many drugs and methods get patented if you can make a huge dealing off of finding such processes.
The difference between an S and a T is based on whether phosphates are added to the nucleic acid. For instance, if the structure only contains the sugar and the nitrogenous base this is referred to as a nucleoside. If it has phosphates of any number attached it becomes a nucleotide.
Stella, V. J., & Rajewski, R. A. (2020). Sulfobutylether-β-cyclodextrin. International journal of pharmaceutics, 583, 119396. https://doi.org/10.1016/j.ijpharm.2020.119396
Schooley, R. T., Carlin, A. F., Beadle, J. R., Valiaeva, N., Zhang, X. Q., Clark, A. E., McMillan, R. E., Leibel, S. L., McVicar, R. N., Xie, J., Garretson, A. F., Smith, V. I., Murphy, J., & Hostetler, K. Y. (2021). Rethinking Remdesivir: Synthesis, Antiviral Activity, and Pharmacokinetics of Oral Lipid Prodrugs. Antimicrobial agents and chemotherapy, 65(10), e0115521. https://doi.org/10.1128/AAC.01155-21
Deb, S., & Reeves, A. A. (2021). Simulation of Remdesivir Pharmacokinetics and Its Drug Interactions. Journal of pharmacy & pharmaceutical sciences : a publication of the Canadian Society for Pharmaceutical Sciences, Societe canadienne des sciences pharmaceutiques, 24, 277–291. https://doi.org/10.18433/jpps32011
When the new ideas are not rewarded, (or they are punished) people keep treading down the same old path, and repeated failure. Government has entrenched this model of the definition of insanity.
This information was actually very interesting. There’s a lot people probably don’t understand about drug design. And now I understand why I add mustard to my salad dressing - to marry the ACV and EVOO.