


An Introduction to Pharmacology
A mini lesson on drug design and pharmaceuticals.

I’ve always considered being a teacher and was considering going to graduate school to become a professor. Of course, life had a different idea in mind. Although I didn’t become a teacher I still love the idea of learning and spreading knowledge. Many of my newsletters have been made with the assumption that my readers will understand a lot of the information which was not the proper approach. So instead, I’ve decided to create a separate newsletter section (for now called Mini Lessons) that will serve to hold mini lessons on various science topics and will serve as a place for people to gain general information. With all of these posts I will include an Applied Knowledge section where I will ask some basic questions. The answers to these questions will be included in a later paid-only post. However, feel free to ask questions if there are some topics that are confusing.
Now, I have a slight confession to make: I find clinical trials rather boring. Yes, yes, it’s a bit heretical to say such a thing! Clinical trials serve many purposes and they tell us whether a drug is effective on humans for specific diseases or if they may be come deadly. However, in most instances clinical trials just show that a drug works but they don’t fully explain how or why a drug ends up being effective, which I find far more interesting. How a drug circulates around the body, which organs are the main targets, and which proteins drugs may bind to all fall upstream of the clinical presentations of an effective therapeutic.
The field of science that focuses on drugs and their interactions and effects on living organisms is referred to as pharmacology. Pharmacology serves as the cornerstone for modern day medicine and drug discovery. It’s an interdisciplinary approach that combines concepts from various fields such as biochemistry, medicinal chemistry, physics, computer science, and mathematics that tries to answer the “how” and “why” about drug interactions.
The history of pharmacology is a little muddy. The first instances of pharmacology in action may draw its roots back to the early 1800’s where Francois Magendie provided a plant, nux vomica which causes muscle contractions, to dogs. Magendie found that the site of the active compound- strychnine- was the spinal chord and caused convulsions in these dogs.

Now, you may be looking at this and wondering why pharmacology doesn’t date back many centuries, as it is very well-known that many of our ancestors turned to herbs, trees, and other forms of vegetation to treat ailments. We could refer to the early 1800’s as the start of modern pharmacology, but we can also make an argument that pharmacology is intended to answer the “how” and “why” to drug interactions, and not “what” a drug does. In that sense, the lack of scientific methods from centuries prior would have limited the knowledge of herbs to what they are used to treat, and not how these drugs operate within our bodies. This concept is not fully concrete but keep in mind that there will be different interpretations about the history of pharmacology depending on where you look.
More historical information on the study of pharmacology can be found on the American Chemical Society website.
Pharmacology examines the effects of drugs, which can be loosely defined as exogenous (foreign) compounds to the body that elicit physiological effects. This includes synthetic, pharmaceutical compounds as well as compounds sourced from plants and animals. Because the effects of compounds depend upon how much one ingests (dosage), and considering that everything has a toxic dosage, the field of pharmacology greatly overlaps with the field of toxicology which studies how compounds cause deleterious effects on our bodies.
Pharmacology can be divided into two main categories; pharmacodynamics and pharmacokinetics. Each division will get their own explanation further on, but they tend to get mixed up. In short, pharmacodynamics looks at what drugs specifically bind to within our bodies while pharmacokinetics looks at how and where a drug ends up after being administered.

Pharmacodynamics: What a Drug Does to the Body
We take supplements and therapeutic drugs with the intent that the drug will eventually bind to a some protein within our bodies that ends up benefiting us. Pharmacodynamics examines the biochemical, physiological, and molecular effects of a drug on the body. This entails the interaction of drugs with many of our cellular machinery which alters their effects. Essentially, pharmacodynamics looks at the actual dynamics of a drug binding to specific proteins and causing an effect.
Most of these interactions focus on drug-receptor interactions, although many drug interactions are not receptor dependent. For example, antivirals and antibiotics don’t necessarily need to bind to receptors, and instead bind to viral and bacterial enzymes to elicit their actions. In fact, enzymes1 are one of the biggest targets of drugs.

Receptors are proteins found both inside and on the outer membrane of our cells that are involved in chemical signaling either within or between cells. Compounds that bind to these receptors are called ligands and include drugs, neurotransmitters, and ions. The interaction between a drug and a receptor can either increase or decrease chemical signaling (taken from Currie, G.):
Receptors are proteins (macromolecules)—inside or on the surface of cells—that mediate drug activity (1–3). Receptors respond to specific neurotransmitters, hormones, antigens, chemicals, or substances. A chemical (ligand) binds to a specific site (receptor) and triggers a response (signal) in the cells (1–3). The intracellular changes initiated by the ligand–receptor complex can be through direct or indirect action; however, the ligand generally functions as either an agonist or an antagonist (1–3). An agonist will mimic the endogenous ligand to produce a similar response (e.g., morphine is an agonist for opioid receptors), whereas an antagonist blocks the usual ligand and thus inhibits the physiologic response (e.g., naloxone is an antagonist for opioid receptors) (Fig. 2) (1–3). A more detailed explanation of terms regarding drug action is presented below (1,2).
As stated above, a drug that evokes a similar response to our own endogenous ligands are called agonists. Examples include opioid agonists such as morphine and heroine which bind to opioid receptors and elicit an analgesic (pain reducing) effect. Sometimes drugs cannot elicit the same effects as endogenous ligands, and instead of causing an all-or-nothing effect a drug may cause an attenuated stimulatory effect. Such drugs are considered partial agonists. However, if a drug intentionally blocks the activity of a receptor the drug is considered an antagonist. A common example of a receptor antagonist are ACE-II antagonists that block the activity of ACE-II receptors in order to reduce blood pressure.
Because drugs need to compete against endogenous ligands to bind to receptors, a drug’s effect is considered to be dose-dependent. In theory, higher levels of a drug would mean that more drugs can compete to binding to a receptor and cause the desired outcome. However, we know full well that you can’t provide as much of a drug as you want. At some point the adverse cytotoxic effects of a drug outweigh any benefit. The proper dose of a drug tries to maximize the effect while minimizing the harm.
Pharmacokinetics: What our Bodies do to Drugs
Just as important is the activity of a drug when administered, the way that a drug is administered and how it makes its way around our bodies are just as important. Pharmacokinetics examines how drugs make their way into our bodies, how they get to the intended site, and how our body gets rid of the drug after it has fulfilled its duties. Pharmacokinetics follows the acronym LADME with each letter describing a different contributing factor to drug delivery. The acronym stands for the following:
Liberation2
Absorption
Distribution
Metabolism
Excretion

Liberation
Liberation refers specifically to drugs that require a carrier agent delivery system. In order to become bioavailable a drug needs to be released from a delivery system, hopefully at the intended site. A good example will be the mRNA and DNA vaccines, which require that the genetic material (the drug) be properly released from the lipid nanoparticle or adenovirus vector into host cells. Another example would be Remdesivir, which uses a carrier molecule to help emulsify the drug due to its high hydrophobic nature and carry it into the bloodstream. If a drug gets too stuck within the carrier agent it will not be able to elicit the proper response.
Absorption
Absorption describes how a drug, based on how it gets administered, enters into the body. Absorption heavily depends upon the structure of the molecule, and actually dictates how a drug is administered as certain routes are far better than others depending on what the drug is. Some of the important factors for absorption include the following:
Solubility: Whether a molecule prefers water or fat affects how it enters into the body. The idea here is that the route should allow the drug to be the most bioavailable. Most drugs must balance hydrophilicity and lipophilicity in order to circulate within the body and properly pass through the lipid membrane of cells. In general, most drugs are able to be administered orally which is the least invasive approach. However, sometimes a drugs solubility makes it difficult to be administered orally and therefore must find another another route of entry into the body. For example, compounds such as testosterone used for hormonal therapy are injected into fatty regions of the body such as the glutes. Here, they can become readily distributed in fatty tissues and slowly released into the body.
First-Pass Effect: You don’t want a drug to become metabolized before it can act. This effect is known as the first-pass effect and it explains how drugs may become metabolized/inactivated before eliciting any of its intended effects. The main organ of concern is the liver as it metabolizes nearly all of the compounds we ingest on a daily basis. Another example is the stomach where stomach acids may accelerate the breakdown of a drug thereby inactivating it before it can reach the intestines and pass into the bloodstream. In order to avoid the first pass effect a drug is usually administered in a manner that allows for direct entry into the circulatory system. A good example of this is the drug Nitroglycerin which is used to treat anginal chest pain. Because the drug needs to reach the heart it cannot be inactivated by the stomach or liver enzymes. Therefore, nitroglycerin is administered sublingually (under the tongue) where it passes through the thinner membranes and enters into the bloodstream. A common way of avoiding the first-pass effect is to provide a drug through direct parenteral injection, usually intravenously. This route bypasses the liver and stomach and ensures that more of the drug is bioavailable. It’s worth noting that although many drugs are prescribed orally, many of these drugs have very low bioavailability and a majority of a drug undergoes first-pass metabolism3. Refer to Table 1 in the Curie, G. article for a list of drug administrations routes along with their advantages/disadvantages.
When you look at a how a drug is administered keep in mind that the route of administration matters and can tell you a lot about the properties of the drug.
Distribution
As soon as a drug enters the body it has to reach the intended organs with sufficient concentration. In most cases the drug enters into the bloodstream and begins circulating around the body until it reaches the desired tissues. This term is called distribution. It seems like a simple concept but keep in mind that many barriers prevent a drug from reaching its proper destination.
For one, many proteins within the blood such as albumin and other glycoproteins may bind to the drug. In some cases this helps circulate the drug around the body, especially if the drug may be too lipophilic and requires some assistance. However, if the drug binds too effectively to these plasma proteins they will not be available to properly function. The ability of a drug to be available is referred to as bioavailability. Therefore, a balance between free, bioavailable drugs and protein-bound drugs is necessary if a drug is to function properly. In many cases a drug has an equilibrium point between free drugs and protein-bound drugs. At higher drug concentrations plasma proteins may become saturated meaning that any drug provided at these higher concentrations will be bioavailable as plasma proteins will not take up any more of the drugs. At lower concentrations equilibrium may not favor protein-drug binding. In this case drugs caught up in plasma proteins may be released into the bloodstream and become available.
Both scenarios need to be kept in mind and dictate the dosage regimen used for medications. Some drugs may suddenly be released from plasma proteins and cause a sudden spike in free drug which may become detrimental. Also, if two drugs compete for the same plasma proteins then the free/bound drug equilibrium will be disturbed and the drug dosage may become inconsistent. For example, aspirin and warfarin bind to similar plasma proteins, resulting in some controversies as to whether they should be administered together.
A few more things to note about distribution:
Because distribution mainly occurs in the circulatory system (bloodstream) blood flow greatly alters how quickly and how much of a drug circulates through the body.
Capillary permeability alters how much of a drug moves form the bloodstream into tissues and organs. The blood-brain barrier is very impermeable and makes it difficult for drugs to pass into the central nervous system. The blood-brain barrier serves as one of the largest barriers to overcome in the development of neurodegenerative therapies.
Metabolism
Metabolism refers to the set of chemical reactions that modifies or breaks down a drug into new products called metabolites. The main site of drug metabolism is the liver although the stomach, kidneys, and gastrointestinal tract may engage in metabolism as well. Metabolism serves a few main purposes in drug design:
Metabolism should prevent a drug from continuously recirculating. More lipophilic drugs may bypass kidney excretion and may become reabsorbed into the body and recirculated. The inability to properly eliminate a drug may end up causing adverse reactions over a prolonged period of time. Therefore, a drug should be designed with the ability to be metabolized and properly removed, which usually involves the drug being metabolized into a more water soluble metabolite that can be readily removed from the kidneys. In some cases you may want a drug to stay within the bloodstream in which case you may hinder its metabolism. A good example of this is protease inhibitors used for HIV therapy and the recent anti-SARS-COV2 drug PAXLOVID. HIV protease inhibitors such as Lopinavir are administered with a Cytochrome P450 enzyme inhibitor such as Ritonavir that prevents the breakdown of Lopinavir. This allows Lopinavir to circulate further with the intent of increasing the levels within the bloodstream. PAXLOVID contains both the protease inhibitor Nirmatrelvir and the P450 inhibitor Ritonavir.
Metabolism may be necessary to create an active form of a drug. Some drugs may be easily recognized by enzymes which may break down a drug prematurely. To avoid this from happening a drug may be formulated in a prodrug form which undergoes an enzymatic reaction to become the active drug. This usually occurs with nucleoside analogues such as Molnupiravir, Remdesivir, and Tenofovir which contain a protective group that prevents nucleases from breaking down these drugs prematurely. The protective group is usually removed by liver enzymes and produces an active form of these nucleoside analogues.

Metabolism should inactivate a drug or make it less effective without producing a more toxic product. One of the most important aspects of drug design is whether a drug may be metabolized to become more active and possibly toxic. Ideally, drugs become less active or become completely inactivated by metabolism. However, in some cases the metabolite may be active and exhibit its own toxicity, usually by forming reactive oxygen species or free radicals, or sometimes by binding to cellular proteins and enzymes and causing cell death. A good example is Paracetamol, or better known as Acetaminophen. Although Acetaminophen is considered relatively safe at small doses, prolonged usage may cause one of the metabolites, N-acetyl-p-benzoquinonimine (NAPQI) to bind to liver enzymes and cause hepatic cell death.

For several years the concept of active metabolites has been overlooked in pharmacology leading to many drugs that turned out to be unintentionally toxic. As the field progressed further and more analytical techniques were developed the process of drug metabolism became quintessential in drug design.
Metabolism highlights the fact that it’s not just the compound at hand, but the overall process that should be looked at holistically.
Excretion/Elimination
The last step in pharmacokinetics is the removal of the drug from the body. Usually water-soluble metabolites can be eliminated through the kidneys and urine while more fat soluble molecules may be eliminated through feces. Aside from these two pathways drugs can be eliminated through other processes as well, all depending upon the metabolite. For instance, one of Aspirin’s metabolites is salicylic acid4 which can be released from sweat. If a drug can be broken down into gaseous components it can then be released through the lungs via exhalation.
As we indicated earlier, some drugs may be reabsorbed either through the kidneys or the intestines and recirculate. If the drug has not been metabolized then the effect of the drug may be prolonged.
Although the process of elimination seems simple enough elimination of a drug is based on kinetics and higher order math. We won’t go into details here but know that elimination of a drug is usually determined with kinetic equations.
Each drug has its own elimination rate constant that indicates the rate at which a drug is eliminated from the body. An easy interpretation of this rate is to examine the half-life of a drug, which is the amount of time it takes for half of a drug’s concentration to be eliminated from the body. The half-life measure usually dictates the dosage level and timing of a drug in order to balance out the therapeutic amount with the amount being excreted. Essentially, drugs with shorter half-lives may require higher doses within shorter intervals compared to drugs with longer half-lives, although this depends greatly upon the therapeutic dosage of each individual drug.
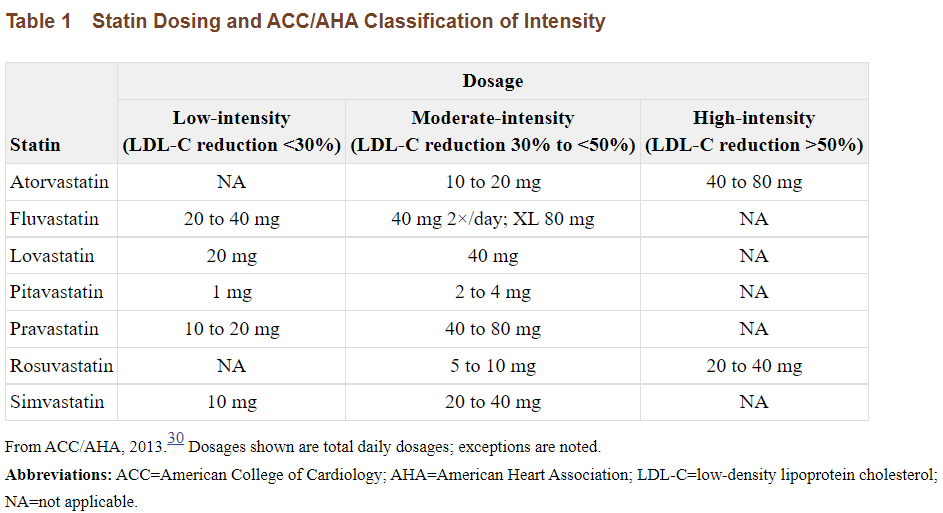
For instance, Penicillin has a suspected half life of around two hours while Atorvastatin, a commonly prescribed statin to reduce cholesterol, has a half-life of around 14 hours. Although the dosing of Penicillin depends upon the type of bacterial infection being treated, we can see how the very short half life affects the dosing regimen compared to typical statins.

Typical half-life measures are based upon a drug’s dosage which may not provide a clear picture to the biochemical activities that go on within the body. Remember that plasma proteins and other physiological circumstances may alter the bioavailability of a drug. Under such circumstances it may be more accurate to use another value called the terminal half-life. This half-life measures the amount of time it takes for half of the plasma concentration to decrease after it has reached pseudo-equilibrium. We won’t go into detail in regards to this measure, but it’s worth noting that the terminal half-life of drugs are extremely long and may last upwards of several months after a drug has been administered.
Lastly, several factors alter the way a drug is eliminated. Most notably impaired liver and kidney function attribute to impaired drug elimination. Diets may also alter drug excretion by upregulating or downregulating enzymes depending upon nutrients that someone consumes. Water can increase urinary elimination as well.
Things to Consider
When we take medications we don’t think too much about the drug. In most cases, we may have a headache and decide to take Acetaminophen or we have a stomach ache and take Pepto-Bismol. We may be prescribed antibiotics for an infection or statins to help lower cholesterol. In many of these circumstances we know that the drug is intended to treat something, but we may not stop and think about what a drug does to cause such an effect or why our symptoms become alleviated.
Pharmacology has developed as a field that tries to understand the multifaceted nature of both drug design and drug administration. It’s a holistic approach that draws from many fields in order broaden the relationship between us as humans and drugs as therapeutic agents.
It’s a concept we tend to overlook leaving us blind and unknowing to the drugs we consume on a daily basis. As COVID has led more people to become skeptical of the medical industry, it’s become ever more important that people understand how drugs operate and why drugs undergo the developmental processes that they do.
So if you haven’t taken your daily supplements or medications yet, take a look at what you have with through the lens of pharmacology. Consider how your drug circulates in your body, how its metabolized, and how it is removed from the body (pharmacokinetics). Also, consider what enzymes and receptors are targeted by your drug and how this leads to the intended therapeutic effect (pharmacodynamics).
Applied Knowledge
Using the information provided and other information that can be found on this Substack let’s try a brief activity to see if you can apply the information you have learned and answer the following questions:

Although questions 1, 2, and 4 are the most pertinent questions, it may be more important to see if you can source information and gain a general understanding of the drugs that you take. So if you cannot find specific answers try to gather any information you can- there’s no harm in learning! Let’s look more specifically at the following drugs:
Acetaminophen
Statins such as Atorvastatin
ACEII Inhibitors such as Losartan- part of the answer for this class of drugs is in its name
Correction 4/5/2022: Losartan DOES NOT target ACEII, but rather targets another receptor closely named called Angiotensin-II Receptor. They sound very similar and follow similar pathways, although they are quite different. Apologies for making such an egregious mix-up!
Molnupiravir
Quercetin
The answers will be posted in a paid-only post, although please feel free to ask questions for clarification and for additional information.
Suggested Reading
Currie, G. M. Pharmacology, part 1: Introduction to pharmacology and Pharmacodynamics.5 Journal of Nuclear Medicine Technology Available at: https://tech.snmjournals.org/content/46/2/81.
Currie, G. M. Pharmacology, part 2: Introduction to pharmacokinetics. Journal of Nuclear Medicine Technology Available at: https://tech.snmjournals.org/content/46/3/221.
Flower R. J. (2013). Pharmacology 2.0.6 British journal of clinical pharmacology, 76(5), 625–629. https://doi.org/10.1111/bcp.12088
Principles of Pharmacology - Study Guide.7 Principles of Pharmacology Study Guide Available at: https://open.lib.umn.edu/pharmacology/.
Sobur, K. Introduction to pharmacology introduction to pharmacology. 8Academia.edu (2016). Available at: https://www.academia.edu/25527671/Introduction_to_Pharmacology_Introduction_to_Pharmacology.
Scheindlin, S. A Brief History of Pharmacology. Modern Drug Discovery Available at: https://pubsapp.acs.org/subscribe/archive/mdd/v04/i05/html/05timeline.html.
Additional Citations:
Zhang, Z., & Tang, W. (2018). Drug metabolism in drug discovery and development. Acta pharmaceutica Sinica. B, 8(5), 721–732. https://doi.org/10.1016/j.apsb.2018.04.003
Attia S. M. (2010). Deleterious effects of reactive metabolites. Oxidative medicine and cellular longevity, 3(4), 238–253. https://doi.org/10.4161/oxim.3.4.13246
I love this. Thank you. Why are ace inhibitors not used as a treatment for covid since the spike molecule binds to ace receptors? As an RN I often looked up drugs for safety information but found the description of the drug included the modifier that the mechanism of action was unknown. What effect does the stomach play in the breakdown availability of oral medications. Weight loss surgery, acid blockers ect?
This was more interesting than I thought it would be, thank you! 👍🏽💕