


Missing the Immune System for the Antibodies
A few ramblings about why COVID antibody studies don't show the full scope of immunology.

Minor Correction: In a recent post I remarked that Bamlanivimab lost effectiveness during the Alpha wave of the pandemic. That was incorrect, as it was the Beta and Gamma waves were responsible for Bamlanivimab not working. The previous article has been corrected.
Edit/Correction: A correction has been added to Footnote 13 with respect to graft vs. host disease. I used the term too broadly when the term refers specifically to bone marrow transplants. For organ transplants, it is generally referred to as organ rejection. Thanks to Clarisse for clarifying. A paragraph from her comment was added as the correction.
The complex system that is the immune system
*from an amateur’s perspective
Given how much we’ve gone through with COVID, we may consider ourselves to be fairly knowledgeable on a few topics such as the immune system. In some sense, many people became novice scientists overnight. There’s nothing wrong with that, and in fact it should be something heavily encouraged rather than what is normally seen within the mainstream scientific press who tends to dissuade people from doing their own research, and would instead invest time in misinformation and disinformation campaigns rather than, well, informing these people.

I will always be reminded of a chyron on a local news station which played during a segment about convalescent plasma therapy, in which the chyron displayed something along the lines of, “Antibodies help fight viruses.”
It always rubbed me the wrong way, mostly because it felt like something so obvious yet someone in that news station thought it was a perfectly “sciency” thing to say, as if to say the common folk don’t know what antibodies are, or what they do (we’ll dabble into this idea later on).
This is made even more strange when you realize that everything about immunity has been done through the purview of antibodies. Most antibody studies are based around collecting blood, spinning it down to isolate the antibodies, then shoving those antibodies in which the antigen and seeing if they kiss and stick together.
If a neutralization assay is more to your liking, you take those antibodies, shove them in with the virus or a pseudovirus, then provide some cells serving as a seductress to readily get infected by the virus. If the antibodies neutralize the virus, the cells remain (relatively) unscathed; if not, the cells are targeted and die off.
And that’s generally it. Well, not generally it, but you can see why many of these studies fail at capturing all of what’s going on with your immune system. We’ve mostly used antibodies as some be all/ end all measure for our immune system, and yet that can’t be further from the truth.
So this (albeit scatterbrained rant) will explain why most antibody studies generally miss out on the broader picture and only provide a very narrow glimpse into the world of immunology.
Remember that our immune system is comprised of both innate and adaptive immunity, yet we generally don’t discuss anything remotely associate with innate immunity within the context of COVID, even though the innate immune system is one of the first lines of defense at a critical viral entry point (i.e. the nose, mouth, and other mucus membranes).
A little bit on the innate immune system
The innate immune system is one of the first immunological point of contact for pathogens. This immune system is considered to be broad because it must deal with a host of different bacteria and viruses, and therefore acts slightly indiscriminately.
Here are some innate immune cells that help fight diseases1:

We generally don’t look too deeply into what’s occurring in important infection zones such as the nose where the innate immune system fights off the virus as soon as we inhale the virions. Children are considered to be generally protected through their innate immune system, but even adults generally have a good degree of protection as well. It’s generally later on in our life do we run into a few problems with our innate immune system.
This is a very broad issue, such that even within the realm of immunity the focus has been solely on the adaptive immune response2, and yet it is the innate immune response that gets first strike at infectious agents. It’s the complimentary nature of both the innate and adaptive immune response that keeps us alive, and in fact we may argue that a proper, functioning innate immune system may serve us better than to focus solely on the behaviors of the adaptive immune system.
Even though research into innate immunity in relation to SARS-COV2 has been occurring, the general lack of discussion in the mainstream and alternative outlets may suggest that many people are completely oblivious to the significance of our innate immune systems.
As an aside, there has generally been this idea that the innate immune system is not trainable, and a preprint indicating that remarking that it [trained innate immunity] was quite possible with the vaccines began to circulate around Substack and stired up a bit of an uproar with the general phrase, “this shouldn’t be happening”, popping up a few times.
The only problem is, recent evidence appears to suggest that the innate immune system is actually trainable- go figure!
Here’s the abstract from a review article by Netea, M. G.3 detailing this trained immunity:
The inability of innate immunity to build an immunological memory is considered a main difference with adaptive immunity. This concept has been challenged by studies in plants, invertebrates and mammals. Recently, a paradigm shift in our understanding host defence has been triggered by the mounting evidence for innate immune memory, leading to increased responses to secondary infections. Important differences between the cell populations and the molecular mechanisms exist between the adaptive traits of innate host defence on the one hand and immunological memory of adaptive immunity on the other hand. The lasting state of enhanced innate immunity termed ‘trained immunity’ is mediated by prototypical innate immune cells such as natural killer cells and monocytes/macrophages. It provides protection against reinfection in a T/B-cell-independent manner, with both specific mechanisms and nonspecific epigenetic reprogramming mediating these effects. This concept represents a paradigm change in immunity, and its putative role in resistance to reinfection may represent the next step in the design of future vaccines.
I personally was not aware of this ability for the innate immunity to be trained, as well as many others who were not aware, but it’s likely because this concept has only been discovered in recent years.
It should remind people that science has not figured everything out yet-there’s still many things that are yet to be discovered, and there’s likely to be many things that were once part of the consensus that may be modified as new evidence emerges. So be careful to assume that things we are seeing now are unique only to the current situation rather than something that science has yet to fully elucidate.
A good bit on the adaptive immune system
Ironically, although the adaptive immune system has been the general focus of COVID via rampant vaccination campaigns, there’s been a general oversight in other facets of the adaptive immune system for specifically the behaviors of antibodies.
For example, if we use this simplified example of the adaptive immune system you can see where nearly all of the focus has been:

Now, let’s be clear here: I am very bad at immunology. This tends not to be my wheelhouse unlike many others here on Substack. However, I can still criticize the sparse investigation into other adaptive immune pathways when many studies stare at antibodies and extrapolate information from just that alone.
Generally, neutralization assays tell you whether things are sticking better or worse than they stuck before (reductive, but many of these assays are intended to see if antibodies are binding well to the virus).
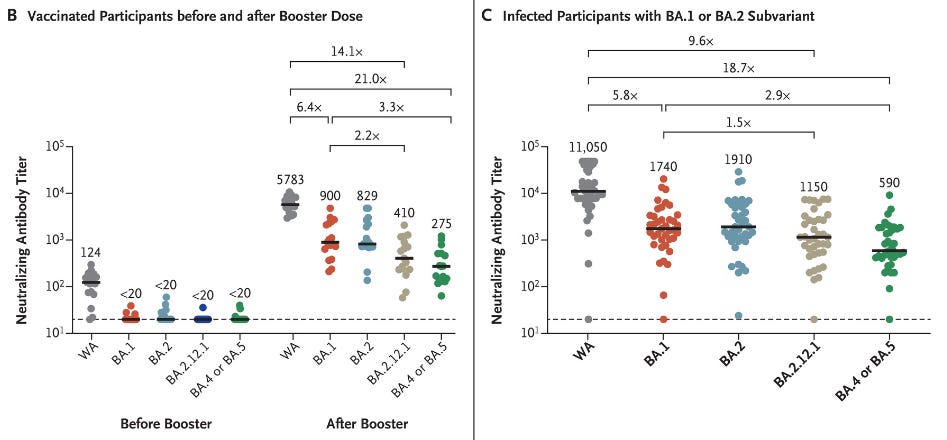
Let’s look at the neutralization assay from Hachmann, et. al.4 from Wednesday’s post which compared neutralization antibody titers before/after a booster, as well as in those who were infected with prior Omicron strains:
When looking at these figures try and see what exactly you can extract from the information.
Well, we know that neutralization capabilities reduced across the different Omicron strains. The general question is, how is this happening? Usually studies such as these don’t tell us why exactly there is a loss of neutralization, and most studies usually don’t refer to the mutations as an explanation for the loss of neutralization.
Also, when looking at the above neutralization antibody titers keep in mind that the results on the left are for 27 participants before and after their booster dose. The one on the right is really difficult to parse because those results are based solely on infection with Ba.1 and Ba.2 with many participants having either been provided different vaccines or on different dosing regimens (refer to Table S1, COVID-19 vaccination history in the Supplementary Appendix for additional information).
The researchers make this comment in regards to the results:
These data show that the BA.2.12.1, BA.4, and BA.5 subvariants substantially escape neutralizing antibodies induced by both vaccination and infection. Moreover, neutralizing antibody titers against the BA.4 or BA.5 subvariant and (to a lesser extent) against the BA.2.12.1 subvariant were lower than titers against the BA.1 and BA.2 subvariants, which suggests that the SARS-CoV-2 omicron variant has continued to evolve with increasing neutralization escape. These findings provide immunologic context for the current surges caused by the BA.2.12.1, BA.4, and BA.5 subvariants in populations with high frequencies of vaccination and BA.1 or BA.2 infection.
The opportune word here is likely “immunological”, which generally may focus on the behaviors of antibodies and immune cells. However, it doesn’t tell us anything about mutations in the spike protein and how those mutations prevent antibodies from sticking. Which mutations are causing antibodies not to stick, and which antibodies are not sticking because they require binding to regions that have now mutated?
This relationship between immunology and virology is typically not examined, so how do we tie these two together?
The answer is a pretty obvious one- we can look to monoclonal antibodies as a way to tie antibodies and antigen together.
Monoclonal antibodies as a reflection for our immune system
Right before Omicron there was a generally huge push for monoclonals on one side of the COVID ideological spectrum. It’s a shame that ideological lines continue to be drawn as it relates to COVID therapeutics, but monoclonals generally ran counter to the COVID vaccine narrative. Many people on Twitter who were fully pro-vax were labelling monoclonal therapies as experimental therapeutics.
It’s pretty funny, because it generally indicated a lack of understand of monoclonal antibodies.
Monoclonal antibodies are isolated from people who have been previously infected, so we can generally assume that they are the same (or at least similar) antibodies that our own bodies produce to fight an infection. I wrote a bit about monoclonals back in September, although some of the information may be out of date (such as my assumptions about Sotrovimab).

Although a few modifications may be made5, monoclonal antibodies in use should in theory overlap with our very own antibodies, and they should tell us a bit about how some of our antibodies may stop binding due to various mutations.
Let’s take Eli Lilly’s monoclonal Bamlanivimab. Bamlanivimab is a monoclonal antibody that binds to the RBD of the spike protein. Unique to Bamlanivimab is that this antibody is able to bind to the spike in both its open and closed conformations.
Bamlanivimab (LY-CoV555) showed effectiveness against the Alpha variant, yet when Beta and Gamma arrived Bamlanivimab stopped being effective. At the same time there were reports that Beta may escape immunity, and so that should clue us in to drawing a few parallels.
So what made Bamlanivimab stop working?
One of the key amino acids for Bamlanivimab was the E484 amino acid residue within the RBD of the spike protein.
We can see that from prior studies, including this 3D model of atomic interactions from Jones, et. al.6:
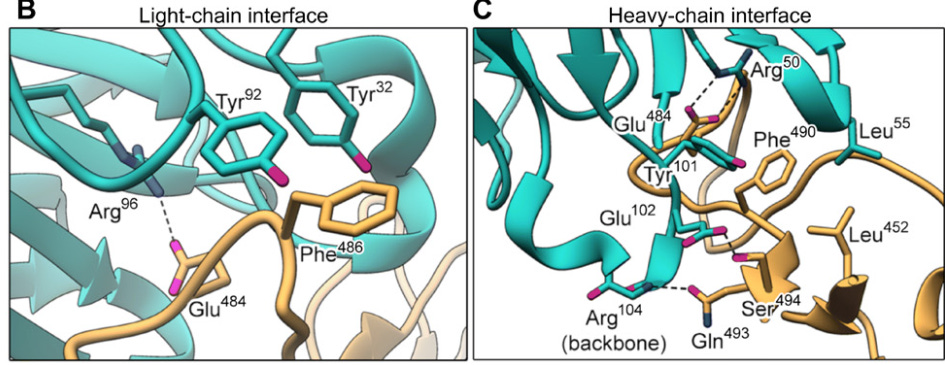
In the 3D model above Bamlanivimab is in cyan while the RBD of the spike is shown in mustard yellow. In both the left and right interfaces, we can see that key binding motifs include the interaction between Arginine Residues (Arg96 on left, Arg50 on right) of Bamlanivimab and the E484 (Glu484) amino acid residue of the spike’s RBD.
Remember that Arginine is a positively charged amino acid while Glutamic Acid is a negatively charged amino acid. Here, both hydrogen bonding and ionic interactions cause favorable binding between the two (i.e. opposites attract).
However, if E484 were to change to something positively charged we may have the opposite effect, such that two positively charged amino acids would cause a repulsive force and reduce the binding affinity between the antibody and the antigen.
The main mutation for the E484 residue is to swap from the negatively charged Glutamic Acid to the positively charged Lysine (K), and in doing so the favorable binding is lost and the antibody (Bamlanivimab) no longer binds.
This E484K mutation is considered to be one of the main drivers of loss of neutralizing antibodies in the variants that carry this mutation.
So in piecing all of this together what does this tell us? It tells us that Bamlanivimab-like antibodies (i.e. our own natural antibodies that would bind to the E484 residue) will not bind to variants that carry an E484 mutation, and within the context of COVID’s viral progression this explanation would explain why there was a drop in neutralization antibodies when going from Alpha to Beta- Bamlanivimab, and antibodies that bind similarly to Bamlanivimab, would be greatly affected.
In essence, the loss of binding for Bamlanivimab should be an indication as to what may be going on with our own antibodies.
Just for one more example let’s take a look at Sotrovimab.
Sotrovimab was developed by Vir Biotechnology in partnership with GlaxoSmithKline. Sotrovimab is also a unique monoclonal, because unlike other monoclonals in use Sotrovimab was actually isolated from a patient infected with SARS-COV during the 2003 outbreak and found great success when used against SARS-COV2.
This search for a cross-reactive antibody is crucial, because it comes with a general assumption that an antibody that can target antigenically different spike proteins may target conserved regions. Conserved regions means that these portions of the spike protein are not likely to mutate across different lineages, and therefore can be easily targeted by one antibody7 and remain effective across several variants.
It’s for that reason that, unlike RegenCOV and Eli Lilly’s therapies Sotrovomab held up during the Ba.1 outbreak8.

Unfortunately, that wasn’t the case during the Ba.2 outbreak. Various studies showed that Sotrovimab lost effectiveness against Ba.2, and eventually the FDA removed the authorization for its use in April 2022 due to Ba.2’s dominance.
So what happened between Ba.1 and Ba.2 that led to Sotrovimab no longer working?
First, let’s refer to the mutational lineage from Wednesday:
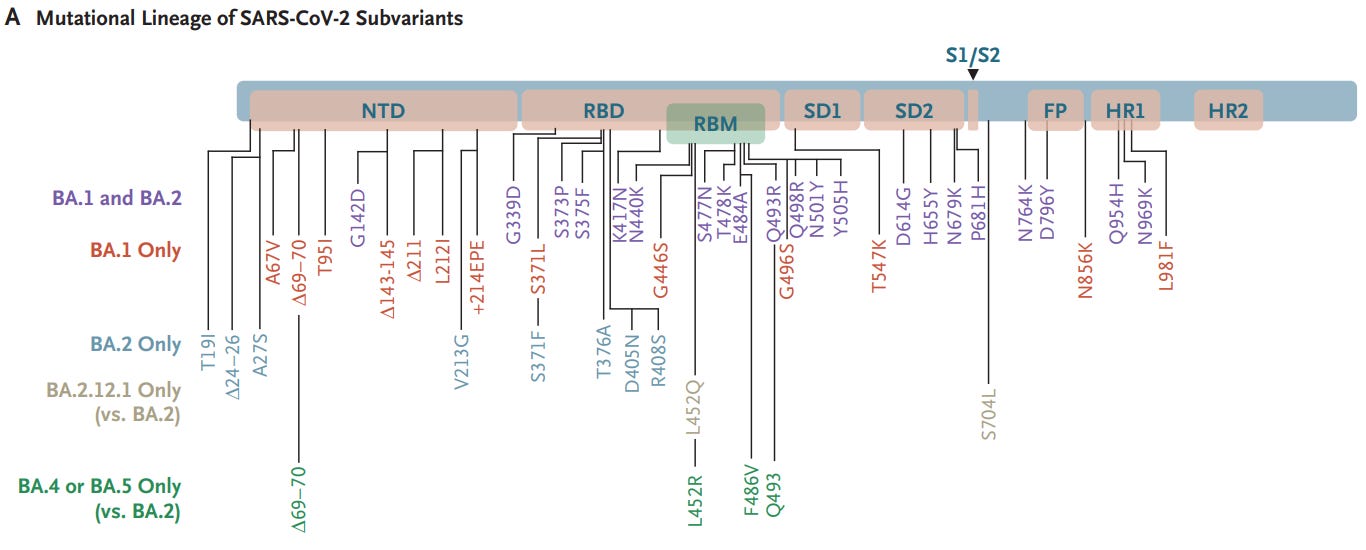
In order to clue us in, we may have to look at the differences in mutations between Ba.1 (in red) and Ba.2 (in light blue). Now, we will use a bit of cheating, but 3 mutations stick out within the RBD- the T376A, D405N, and R408S mutations. These mutations are not seen in any prior variant, and considering that Sotrovimab held up until Ba.2 we may infer that these 3 mutations may be the culprit.
In one study by Zhou, et. al.9 researchers wanted to map various mutations in Ba.2 to antibody neutralization escape. Pseudoviruses containing 4 different point mutations (S371F, T376A, D405N, and R408S- this means that they took the spike protein and only added one of the 4 mutations to test the individual effects of each mutation) were tested, and the results for Sortrovimab were as follows:

We can see that the 4 mutations may be responsible for Sotrovimab’s loss of neutralization. It’s interesting that the S371F appears to have affected Sotrovimab, as the S371L mutation was found in Ba.1, although both are likely to have altered Sotrovimab’s binding capacity. In both instances, the S371F and S371L mutation takes a polar, hydrogen-bonding competent Serine residue and replaces it with a nonpolar Phenylalanine and Leucine, respectively. The loss of hydrogen bonding could be responsible for the decreased neutralization. However, the addition of the T376A, D405N, and R408S mutations may have helped as well, and the collection of mutations may be to blame for the overall loss of neutralization against Ba.2.
These results are rather fascinating, because in this scenario it appears that the mutations are not responsible for direct interactions between Sotrovimab and the spike protein, but apparently the mutations may have changed the conformation of the spike and that itself may be the reason for the loss of binding:
For the most part, the active mutations in BA.1 mapped to the sites of interaction with the antibodies while the additional active mutations in BA.2 (S371F, T376A, D405N and R408S) lie distal to the interaction sites (Figure 4). Presumably, these mutations might affect the conformation of the RBD spike.
Unfortunately, these remarks are more suggestive rather than substantiated- further evidence would be needed to argue that a conformational change may have occurred in Ba.2. The researchers do remark, and through their 3D modeling as well, that the S371F mutation may interact with Sotrovimab in some way.10
Regardless, this alludes to the fact that something has happened due to the above mutations to cause Sotrovimab to no longer bind. This generally explains why Ba.2 was considered to have some level of immune escape- antibodies that may have worked on prior variants, including Ba.1 may not have worked against Ba.2 due to the 4 mutations within the RBD. A few things worth considering when relating our own antibodies to immune escape across different variants.
As an aside, it does raise questions as to the origins of both Ba.1 and Ba.2 and why Ba.2 may carry a mutation within a generally conserved region of the RBD, but we may save that for another day.
Monoclonals allow us a window into what occurs within our own bodies on a daily basis, except they don’t operate alone. All of us may have a sprinkling of Sotrovimab, a dash of Bamlanivimab, a few hints of Casirivimab and Imdevimab, and maybe some Tixagevimab for good measure. It’s the sum of our antibodies working in tandem, either binding to similar epitopes (Bamlanivimab and Etesevimab) or binding to separate positions (Casirivimab and Imdevimab) or even targeting conserved epitopes that are found across different variants (Sortovimab) that allow us to continue live and fend of diseases11.
So when examining neutralization assays- or any antibody assays for that matter- keep in mind that there’s far more going on that just antibodies sticking to antigens. Think about the types of antibodies and what epitopes they may be targeting. If those epitopes change, how will this affect monoclonal therapies, as well as my own antibodies?
HLA and why (immunological) diversity is our strength
The last topic of discussion here will be something called the Human Leukocyte Antigen (HLA). HLA is generally taught in high school as the major histocompatability complex (MHC), but for us humans we specifically have the HLA.
This review will be very limited due to the scope of HLA and its relationship to adaptive immunity.
Generally, when examining HLA we focus on two separate classes of HLA, with each class having 3 genes that serve as the primary focus:

Class I comprises the A, B, and C genes while Class II generally consists of DR, DQ, and DP.
Class I antigens are generally found on the surface of all nucleated cells while Class II is generally reserved for B lymphocytes and antigen-presenting cells such as dendritic cells and macrophages.
The HLA system is one of the most critical regulators of our immune system as they are responsible for binding to antigens and presenting them to T-cells. They help alert the immune system to an infected cell so that they may be targeted for cell death. They can also carry antigenic peptides to T-cells (called antigenic presentation) so that they can alert them to possible dangers to look out for.
Because of their role in our immunity, you may not be surprised to find out that HLA genes are some of the most diverse genes within the human body (Choo, S. Y.):
The HLA system is known to be the most polymorphic in humans. The HLA polymorphism is not evenly spread throughout the molecule, but is clustered in the antigen-binding groove.2,5,8 Amino acid variations in several regions change the fine shape of the groove and thus alter the peptide-binding specificity of HLA molecules (see below for details).11 The distribution and frequency of HLA antigens vary greatly among different ethnic groups. It has been postulated that this diversity of HLA polymorphism has evolved under unique selective pressure in different geographic areas. This could be related to the role of the HLA molecules in the presentation of prevalent infectious agents in the different areas of the world.
As an example, let’s use this Wikipedia article which suggests that, as of March 2022, there are over 7,400 different HLA-A alleles discovered and about 4,300 different active proteins12 discovered so far.
If you remember from genetics, our chromosomes are paired, which means that we can carry two different alleles for the HLA-A gene on each individual Chromosome 6. If we take that into account, we can see the astronomical level of variations possible.
For example, even within just this one gene we can expect up to 54 million different combinations of HLA alleles (7400^2), or up to 18 million different combinations of proteins (4300^2)13. Factor in the other genes and you can see the different combinations may be almost limitless.
But important to our discussion is what role these differences in HLA genes do to our immune system.
The high variety of HLA genes means that each of us (and our APCs) can present different antigens to our T-cells. A carrier of one HLA-A allele may carry one peptide from the spike while a carrier of another HLA-A allele may target a different peptide from the spike. This means that each of us, individually, will present and respond different to a SARS-COV2 infection. However, this also means that our cells will respond to mutations and viral variants differently as well.
As such, this tells us that there is a deep relationship between T-cell responses as well as the HLA-alleles that we all carry.
Take the Motozono, et. al.14 study from Wednesday, which alluded to examining the effects of the L452R mutation on HLA-restricted cellular immunity.
HLA-restricted immunity, as defined within this article, suggests that HLA-alleles can be highly restrictive in which antigenic peptides they bind to, such that a mutation may cause them to not recognize the peptide anymore, and thus will not alert T-cells to the peptide.
We won’t go too deeply into this study, but the researchers looked specifically at the HLA-A*24 allele, which appears to bind a 9-mer (9 amino acid peptide) peptide that is part of the RBD. This allele appears to predominate in East and Southeast Asia:
A bioinformatic study has suggested that the 9-mer peptide in the RBM, NYNYLYRLF (we designate this peptide “NF9”), which spans 448–456 of the S protein, is the potential epitope presented by HLA-A24 (Kiyotani et al., 2020), an HLA-I allele widely distributed globally and particularly predominant in East and Southeast Asian areas (Table S1). Additionally, three immunological analyses using COVID-19 convalescent samples have shown that the NF9 peptide is an immunodominant epitope presented by HLA-A∗24:02 (Gao et al., 2021; Hu et al., 2020; Kared et al., 2021).
This may explain why, for the most part, Asian countries tended to do fairly well with early COVID variants, although we have no way of knowing who among a given population carry the HLA-A*24 allele.
As the researchers remarked in their study, the L452R mutation likely reduced binding affinity between the HLA-A*24 molecule as well as the 9-mer:
We then assessed the possibility that these two mutations affect the binding affinity of NF9 to HLA-A24 molecules. In silico analyses using five tools (Andreatta and Nielsen, 2016; Karosiene et al., 2012; Lundegaard et al., 2008; Nielsen et al., 2003; Reynisson et al., 2020; Zhang et al., 2009) predicted that the L452R mutation decreases the binding affinity of NF9 to HLA-A24, and four out of the five tools predicted that the Y453F mutation decreases the binding affinity (Figure S1A).
This does come with the caveat that these were in silico studies, but it does allude to the significant dynamics between antibodies, HLA molecules, and antigens, such that the relationship between all 3 is what drives pathogenicity. So not only can a mutation such as the L452R mutation lead to loss of neutralizing antibodies, but it may also reduce cellular-mediated immunity among carriers of the specific allele.
And the last part is what’s important here- the results of the above study are really only applicable to those who carry the HLA-A*24 allele. This is a general problem with these studies with T-cells. Although they provide a broader perspective in regards to adaptive immunity, no one study can take into account the millions upon millions of different combinations of HLA genes possible. This generally means that studies must limit which alleles they look at, and in doing so it makes it nearly impossible to extrapolate or broaden the results of these studies to a wider population.
This is also seen in the Immune Imprinting article from Science15, where one of the studies utilized transgenic mice which carried the DRB1*04:01 allele. The DRB1*04:01 allele has been considered to be associated with more mild/asymptomatic COVID. This allele itself is generally found in Northern latitudes and people of European ancestry.
Now, there’s a ton of issues with this study (we will examine them in another post), however, this study focused solely on this one allele and inferred that T-cell priming may occur depending upon the initial pool of antigens one is exposed to. Even with a few of the problems in that study, the analysis here only extends so far as to those who carry the DRB1*04:01 allele, if at that16.
Unfortunately, what this tells us is that even in our want for adaptive immune studies outside of just looking at antibodies we run into all sorts of problems when we take into account the extremely high variability of HLA genes. It tells us that these types of studies may not be applicable to the individual, and that there may be problems with making such a broad assumption based on these highly selective analyses.
In short, studies generally don’t examine the response by other adaptive immune cells such as T-cells, and when they the scope of the analysis is so narrow it may not be applicable to us as individuals.
The HLA system is one of the most important aspects of our immune system. It alerts the immune system to when our cells are infected, and it tells our immune system what proteins to look out for in case of an invading pathogen. Each of us carry a unique set of HLA molecules, providing each of us our own unique response to an infection. It’s very unlikely that a study can capture the full breath of different combinations, and we should keep in mind such limitations when looking at studies.
Some things to consider…
So at this point you likely became aware that this post ran away from me. I can’t quite remember what my original intent with this post was…
But if there’s anything worth taking away, remember that our immune system is highly complex. There’s many systems and biochemical reactions taking place at any given time that work in tandem to keep us alive.
When we have neutralization or some antibody studies thrown into our faces we may be immediately inclined to look at these studies and say, “see, our immune system is totally shot!” or to extrapolate some vague notion that we are all going to die. It’s far more complicated than that, and there’s a broader issue in which the scientific community has focused so heavily on such a narrow aspect of our immune system.
This generally leaves us ignorant and quick to make assumptions that are likely not based in science.
When looking at studies, remember to keep a thought in the back of your head that asks you if there’s more than what is being presented. Should I be putting all of my immunological eggs into an antibody basket, or should I consider other factors that have yet to be researched?
I think, with time, we may find out that a gross bastardization has happened to the science community in response to the pandemic, but only time will tell if we move away from the antibody heuristic fallacy.
If you enjoyed this post and other works please consider supporting me through a paid Substack subscription or through my Ko-fi. Any bit helps, and it encourages independent creators and journalists outside the mainstream.

Marshall, J. S., Warrington, R., Watson, W., & Kim, H. L. (2018). An introduction to immunology and immunopathology. Allergy, asthma, and clinical immunology : official journal of the Canadian Society of Allergy and Clinical Immunology, 14(Suppl 2), 49. https://doi.org/10.1186/s13223-018-0278-1
I’ve skimmed this article, and this definitely is a very approachable article for those without a deep scientific background. If anyone reads this article please let me know!
Apparently a workshop organized by staff of the NIAID and the NIH was held earlier in the year to discuss innate immunity.
Diamond, M.S., Lambris, J.D., Ting, J.P. et al. Considering innate immune responses in SARS-CoV-2 infection and COVID-19. Nat Rev Immunol (2022). https://doi.org/10.1038/s41577-022-00744-x
Netea M. G. (2013). Training innate immunity: the changing concept of immunological memory in innate host defence. European journal of clinical investigation, 43(8), 881–884. https://doi.org/10.1111/eci.12132
Hachmann, N. P., Miller, J., Collier, A. Y., Ventura, J. D., Yu, J., Rowe, M., Bondzie, E. A., Powers, O., Surve, N., Hall, K., & Barouch, D. H. (2022). Neutralization Escape by SARS-CoV-2 Omicron Subvariants BA.2.12.1, BA.4, and BA.5. The New England journal of medicine, 10.1056/NEJMc2206576. Advance online publication. https://doi.org/10.1056/NEJMc2206576
Researchers may want to make modifications to the Fc (crystallizable fragment) region of an antibody. The Fc is the base of the Y-shaped antibody, and it general serves to signal to B-cells. Modifications may be made to either reduce or enhance signaling depending on which activity is favored.
Jones, B. E., Brown-Augsburger, P. L., Corbett, K. S., Westendorf, K., Davies, J., Cujec, T. P., Wiethoff, C. M., Blackbourne, J. L., Heinz, B. A., Foster, D., Higgs, R. E., Balasubramaniam, D., Wang, L., Zhang, Y., Yang, E. S., Bidshahri, R., Kraft, L., Hwang, Y., Žentelis, S., Jepson, K. R., … Falconer, E. (2021). The neutralizing antibody, LY-CoV555, protects against SARS-CoV-2 infection in nonhuman primates. Science translational medicine, 13(593), eabf1906. https://doi.org/10.1126/scitranslmed.abf1906
For OAS proponents, this should at least allude to the fact that cross-reactive antibodies are inherently desirable for the fact that they can target regions not likely to mutate across different strains of a virus. When analyzing hypotheses be careful to put all of your eggs into one basket. Parse out the information and find the nuances that both substantiate and discredit the hypothesis.
Li, M., Lou, F. & Fan, H. SARS-CoV-2 variant Omicron: currently the most complete “escapee” from neutralization by antibodies and vaccines. Sig Transduct Target Ther 7, 28 (2022). https://doi.org/10.1038/s41392-022-00880-9
Zhou, H., Dcosta, B. M., Landau, N. R., & Tada, T. (2022). Resistance of SARS-CoV-2 Omicron BA.1 and BA.2 Variants to Vaccine-Elicited Sera and Therapeutic Monoclonal Antibodies. Viruses, 14(6), 1334. https://doi.org/10.3390/v14061334

As user sj95 remarked in the comments, SARS-COV2 monoclonal therapy may have benefitted more from using several monoclonals together. There may be logistical or R&D reasons why this did not occur, but as indicated within the literature the use of monoclonals should match the degree of mutation occurring. Considering that SARS-COV2 is mutating at such a high rate, it would be far more favorable to utilize a monoclonal cocktail that provides some level of neutralization. Of course, at that point that monoclonal cocktail may just instead be mimicking our own immune system akin to convalescent plasma therapy.
Many variations within HLA alleles occur within the introns. These intron variations don’t amount to much, as the introns are noncoding regions of our genome. It’s the exons that are translated and express the proteins that we carry. As such, even though many different HLA alleles may differ within their introns it is generally the expressed protein we worry about.
Edit: My information on graft vs host disease and organ transplantation was incorrect. Please refer to the quote below from Clarisse. That section will be kept within the footnote for posterity’s sake, but please note that this is the more accurate description for the disease. Apologies for the incorrect information!:
And, one slight correction to footnote 13 - graft v host disease overwhelmingly refers to bone marrow transplantation and not organ transplants (it very rarely occurs, but when it does it is caused by blood products not being properly flushed from the organ - it's like an organ transplant with a side of bone marrow - the transplanted organ cannot hurt the recipient, but the recipient's immune system can definitely kill the organ - that is simply called graft rejection). Solid organ matching is not precise and many times it is solely based on unacceptable antigens (if they know your body will kill an organ, then you won't get it - otherwise you will be on immunosuppressive drugs for life). The problem with bone marrow transplants is that those cells will inhabit every part of your body and if those cells recognize your body as foreign they will attempt to destroy you - the good news is that immunosuppressive drugs can usually control this until your new bone marrow becomes tolerant of its new host.
This high level of variation is why it is so difficult to find organ and bone marrow matches. Usually many of the genes outlined above need to match between a donor and a recipient (the most important players are the Class I genes, although Class II are considered to be important as well) in order for a transplant to take. Otherwise, the immune system will consider the organ to be different based on the different HLA genes. This can lead to graft vs host disease, in which the mismatch of HLA alleles between the grafted organ/ bone marrow and the host can lead to autoimmune disease and the transplanted organ may be attacked and rejected. Interestingly, since bone marrow transplants are responsible for providing an immunocompromised individual the necessary immune cells to protect themselves, graft vs host disease in a bone marrow transplant recipient would actually lead to the transplanted immune cells attacking the host’s cells due to mismatched HLA molecules expressed on the outside of the host’s cells.
In essence, graft vs host disease in organ transplants will cause the host’s immune system to target the organ, while in bone marrow transplants the transplanted bone marrow will actually attack the host’s own cells. These factors are why it is so difficult to find matches, and even when such a match occurs the prognosis for such a transplant may not be too great because of all of the other confounding factors.
There are a lot of caveats to examining the actual level of protein variations. Generally speaking, the high level of variations are usually due to what’s called molecular typing which is when genes for HLA are sequenced. On a molecular level, researchers are able to discern polymorphic regions. However, on the scale of protein expression many of these variations may not matter. For example, intron variations may be considered negligible, and if these mutations are found within the transmembrane or cytosolic region of the protein they may not be critical to the general behavior of the protein. Also, many ethnic regions generally have certain alleles that dominate over others, likely due to the differences in the pathogenic landscape across the globe. Therefore, although there are likely to be thousands of different alleles across the globe, it is very likely that a majority of the world carries only a few hundred of those possible thousands, which the rest likely being relegated to an extremely small number of the global population.
Motozono, C., Toyoda, M., Zahradnik, J., Saito, A., Nasser, H., Tan, T. S., Ngare, I., Kimura, I., Uriu, K., Kosugi, Y., Yue, Y., Shimizu, R., Ito, J., Torii, S., Yonekawa, A., Shimono, N., Nagasaki, Y., Minami, R., Toya, T., Sekiya, N., … Sato, K. (2021). SARS-CoV-2 spike L452R variant evades cellular immunity and increases infectivity. Cell host & microbe, 29(7), 1124–1136.e11. https://doi.org/10.1016/j.chom.2021.06.006
Reynolds, C. J., Pade, C., Gibbons, J. M., Otter, A. D., Lin, K. M., Muñoz Sandoval, D., Pieper, F. P., Butler, D. K., Liu, S., Joy, G., Forooghi, N., Treibel, T. A., Manisty, C., Moon, J. C., COVIDsortium Investigators‡, COVIDsortium Immune Correlates Network‡, Semper, A., Brooks, T., McKnight, Á., Altmann, D. M., … Moon, J. C. (2022). Immune boosting by B.1.1.529 (Omicron) depends on previous SARS-CoV-2 exposure. Science (New York, N.Y.), eabq1841. Advance online publication. https://doi.org/10.1126/science.abq1841
The researchers do provide predictive binding results for different HLA-A, B, and C alleles within their Supplemental Material. The results of the predictive study can only relate changes in epitopes between Wuhan/Ba.1 and whether this may increase/decrease binding of different alleles.
This is really an excellent overview of the complexities of the immune system. Working in the field of HLA for nearly 25 years I had never actually considered that everyone (based on their HLA) basically presents specific antigens on the surface of their cells. This has got me thinking a lot about what the "vaccine" was hoping to accomplish - I find the cartoons describing what goes on when you are vaccinated with a mRNA shot rather cartoonish (I highly doubt a whole spike protein is gonna make it to the surface of the cell being that it is 3800bp long) - short peptides found in the cell are routinely presented by HLA antigens on the surface of nearly all cells (which are recognized by immune surveillance cells such as dendritic cells or NK cells - cell-mediated immunity is brilliant). I suppose if a vaccine infected cell was lysed then whole spike could get out of the cells. But it does make me wonder if everyone is creating antibodies to the mRNA generated spike according to their unique HLA. I am also wondering about the actual nature of the mRNA generated spike protein considering the use of pseudouridine and codon optimization (exactly how similar is this to an actual SARS-COV-2 spike) - what sort of studies were done to see if this is the case?
On a lighter note I did enjoy the supplemental material (footnote 16) since I was infected with the Wuhan strain and I do know my HLA type - seems I might have some pretty good antibodies (as I was exposed several times to people who were infected with Omicron, but I never got infected again - and I am forced to test every week for my job.)
And, one slight correction to footnote 13 - graft v host disease overwhelmingly refers to bone marrow transplantation and not organ transplants (it very rarely occurs, but when it does it is caused by blood products not being properly flushed from the organ - it's like an organ transplant with a side of bone marrow - the transplanted organ cannot hurt the recipient, but the recipient's immune system can definitely kill the organ - that is simply called graft rejection). Solid organ matching is not precise and many times it is solely based on unacceptable antigens (if they know your body will kill an organ, then you won't get it - otherwise you will be on immunosuppressive drugs for life). The problem with bone marrow transplants is that those cells will inhabit every part of your body and if those cells recognize your body as foreign they will attempt to destroy you - the good news is that immunosuppressive drugs can usually control this until your new bone marrow becomes tolerant of its new host.
It's all really awfully fascinating - I am never not in awe of the immune system! I just wish people would trust their immune systems - and perhaps treat it better! Thank you for delving into this topic!
Great article. I suspect very strongly that our immune systems are still way too complex for the so called "immunology", and therefore "immunologists" cannot yet predict any outcomes that actually matter.
This is okay since science takes time to develop. But I am always suspicious of "immunologists" making sweeping pronouncements, because I suspect that they do not know enough.