


A few notes on Ba.4 and Ba.5
General background on the mutations and the downstream effects.

Correction 7/9/2022: Below I mentioned that Bamlanivimab stopped working during the Alpha wave of COVID, however it actually lost effectiveness during the Beta and Gamma wave. The information has been updated with the correction.
With Ba.4 and Ba.5 reaching the US there’s been another emergence of hysteria from all sides as to what this would mean, and yet in this context there doesn’t appear to be any information as to what mutations within Ba.4 and Ba.5 we should all be concerned about.
So this seems like a good time to review a bit about Ba.4 and Ba.5 and also examine whether this is something akin to vaccine escape or really just a consequence of a typical viral mutation.
In regards to Ba.4 and Ba.5
Before we begin, for those wanting to find additional information on Ba.4 and Ba.5 I suggest this article from Forbes. Strangely, Forbes has been one of the only mainstream outlets out there that actually includes a good bit of the science know-how in their COVID reporting, although I believe that is likely due to the contributor more than the outlet itself.
Also, yesterday Stephanie Brail of Wholistic posted this new article in the NEJM1 with this nice little mutation lineage of Ba.4 and Ba.5 (the Forbes article also contains a mutation lineage as well). Mutation lineages are an outline as to what mutations a subvariant may contain relative to other subvariants. Here, we can see all of the mutations that arose in all of the Omicron subvariants.
Note that, although Ba.4 and Ba.5 are distinct they appear to be clumped together due to both having similar mutations. Ba.4 and Ba.5 appear to have diverged from the Ba.2 lineage of Omicron and so far only appears to have a few mutations of note within the spike protein:
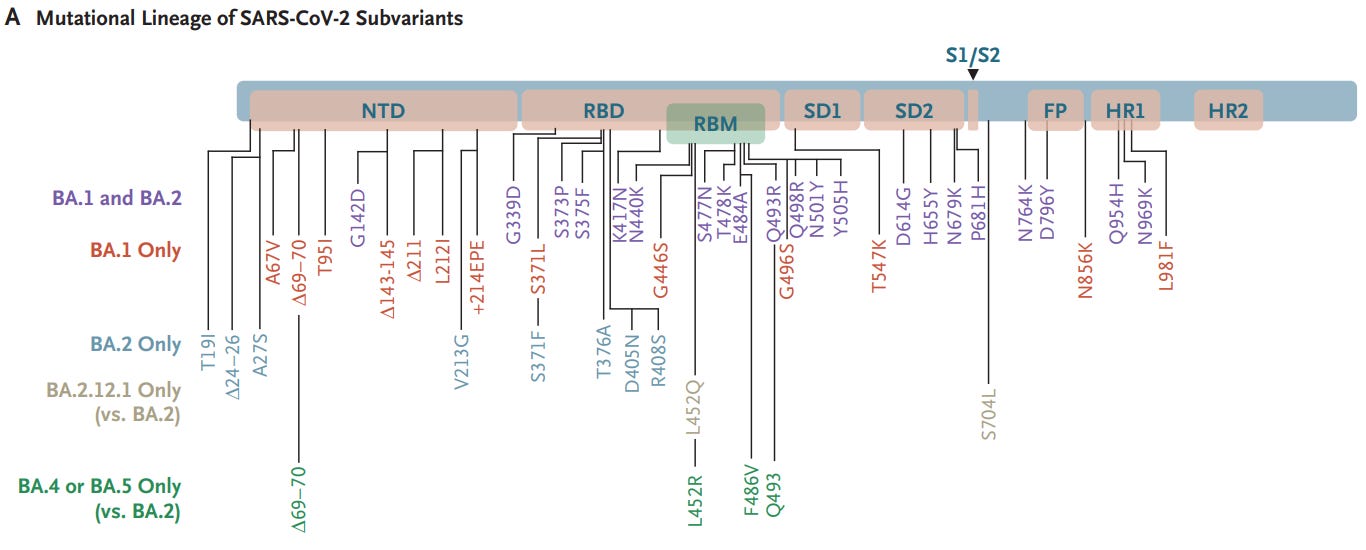
As we can see above there’s about 4 distinct mutations of note here: the addition of the 69-70 deletion that was originally seen with the first strain of Omicron, the L452R mutation, the F486V mutation, and the Q493 reversion. The last one (Q493 reversion) may be the most fascinating of the bunch, as it’s a reversion back to the original glutamine amino acid seen in the prior strains of Wuhan, Alpha, Beta, and Delta.
So we know what mutations differ Ba.4 and Ba.5 from other lineages, the important thing to look at now is to examine what these mutations do and how they may affect neutralizing antibodies.
Before we Begin
In order to look at the relationship between viral mutations and antibodies we first need a bit of a background in amino acids. Amino acids are the building blocks of proteins. Generally there are 20 commonly found amino acids, although there are actually many more that are typically found in the human body.
These 20 amino acids2 are usually grouped by their side chains, which is the structure that differs between all amino acids and confers it’s biochemical properties. These side chains can usually contain a positive charge (as seen with R, H and K below) or they may provide a negative charge (with D and E below). The side chains are the structures shown below the typical amide backbone:

Amino acids derive their functionality from their side chains and how they interact with other amino acids.
Ionic/Electrostatic Interactions
For example, the charged amino acids are an example of opposites attracting, such that positively charged amino acids favor interacting with negatively charged amino acids (R likes D, for instance). This is an example of an ionic interaction and is one of the strongest interactions when it comes to interactions between amino acids.
There are some rare circumstances where two similarly charged amino acids may interact, such as the case for when salt bridges are available. Salt bridges occur when an ionic atom is sandwiched between two amino acids. The amino acids must share a similar charge while the ion must be opposite. An example of this would be a D-Potassium Ion-E salt bridge, such that the two anionic amino acids D and E have a positively charged Potassium Ion (K+) situated between the two amino acids.
This tends to be the most typical viral mutation, which generally swaps one amino acid for another that is ionic in nature, and one that can typically interaction with an oppositely charged amino acid on something such as a receptor.
Hydrogen Bonding
The next favorable interaction is called hydrogen bonding. For the sake of this discussion, we’ll limit hydrogen bonding to discuss amino acid side chains that either contain Oxygen or Nitrogen atoms. Oxygen and Nitrogen sit in an interesting spot on the period table, such that their high electronegativity means that they can both share and receive weak polar bonds made with Hydrogen atoms. When Oxygen or Nitrogen is bonded to a Hydrogen atom, their bond is polar and thus not a full covalent bond. This polarity causes the Hydrogen atom to have a slightly positive charge which can attract other Nitrogen/Oxygen atoms to form their own partial bond to this Hydrogen. Essentially, there’s a shared partnership going on in Hydrogen bond formation.
If A Nitrogen/Oxygen atom is sharing their hydrogen, they are considered to be hydrogen bond donors since they are donating their Hydrogen. Nitrogen/Oxygen atoms who can form bonds to Hydrogen are called hydrogen bond recipients. Although these bond formations are generally weaker than ionic interactions, the overall net effect of many hydrogen bonding interactions occurring is what makes this interaction so strong. It’s essentially the sum of all of its interactions, and the ease of such interactions that makes hydrogen bonds so powerful. When looking at which amino acids can hydrogen bond, remember that hydrogen bonding is possible between ionic amino acids as well as the polar amino acids due to the existence of Oxygen and Nitrogen atoms.
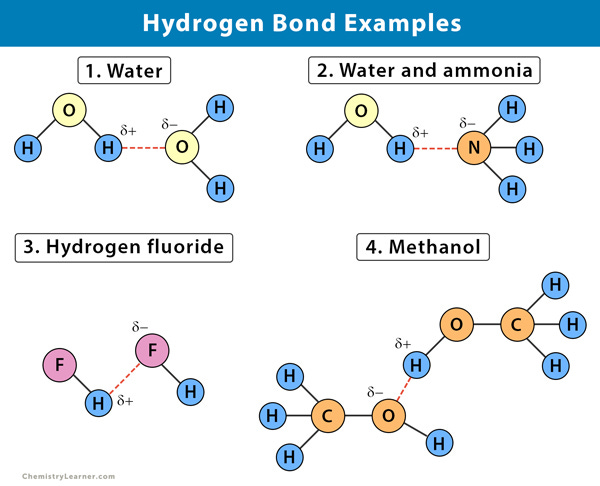
Although there are a few other amino acid interactions, these two are the most important ones for this discussion so we will limit talks to just these two.
Why this Matters
At this point this all may seem trivial are a bit too technical. Remember that science is technical, and in order to understand what is going on one must understand the nuanced nature of science.
Amino acids comprise nearly all living matter- we’re all made up of tons and tons of different proteins, including structural proteins as well as enzymes.
Interactions between amino acids drive biochemical reactions. In the case of viruses, interactions between the amino acids of a virus’ exposed proteins and with our own receptors are what dictates infectivity and pathogenicity of a virus.
Interactions with antibodies- which have paratopes that are amino acid based- and viruses are also dictated by amino acid interactions. Essentially, in order to understand why a virus binds better to a receptor, or why antibodies may lose effectiveness with emerging mutations we must look at the amino acid interaction occurring in order to gain proper insight.
For more, I have written previously on the effects of the N501Y mutation, as well as a bit about the E484K mutation and D614G mutation previously:

Remember to keep all of this in mind when looking at some of the mutations in Ba.4 and Ba.5.
The 69/70 Deletion
So there’s not quite much to discuss in regards to this deletion, mostly because it has appeared in a few of the commonly circulating variants beforehand. This deletion first emerged with Alpha and was found with the first strain of Omicron. However, other subvariants did not have this deletion until the emergence of Ba.4 and Ba.5.
Because this is a deletion, a loss in antibody binding likely stems from not having the proper epitopes for antibodies to target. Therefore, just the absence of these amino acids is likely to prevent antibodies that target this region from binding.
William Haseltine from the Forbes article posited this remark about this deletion:
There is also a deletion of positions 69 and 70 in the N-terminal domain. This is a common mutation found in natural variants and likely knocks out an antibody binding site, meaning this specific mutation could impact overall immune evasion.
Running counter to the immune escape hypothesis above is a study from Meng, et. al.3, in which the researchers suggest that this deletion does not confer immune escape, but likely emerged along with other mutations as a way to balance out reduced binding affinity to the ACEII receptor:
We find that ΔH69/V70 does not significantly reduce the sensitivity of spike to neutralizing antibodies in serum from a group of recovered individuals or binding of multiple mAbs directed against the NTD. In addition, we have shown that repair of ΔH69/V70 does not appreciably alter the potency of NTD antibodies against the B.1.1.7 spike. Thus, the deletion is unlikely to be an immune escape mechanism. Instead, our experimental results demonstrate that ΔH69/V70 is able increase infectivity of the Wuhan-1 D614G spike PV as well as the PV bearing the additional RBD mutations N439K or Y453F, explaining why the deletion is often observed after these immune escape mutations that carry infectivity cost (Motozono et al., 2021; Thomson et al., 2021).
So this deletion may actually serve as some sort of compensatory mechanism for the virus, although it’s worth noting that Omicron does not carry any mutations within the N439 or Y453, although there is a mutation in the 440 position of the original Omicron strain as well as the L452R mutation of the Ba.4 and Ba.5 sublineages.
In short, the 69-70 deletion has appeared in several variants, and although a deletion would be assumed to indicate immune escape, it also suggests a possible mechanism that makes up for loss of binding from other mutations, highlighting the importance of examining multiple factors when assessing what is actually happening. So for now, it may be premature to suggest that this deletion itself is an indication of immune escape or loss of neutralizing antibody activity.
The L452R Mutation
When reading mutations note that the first letter indicates what the original amino acid was (in this case a Leucine). The middle number indicates which amino acid position carries the mutation (the 452nd amino acid). The last letter then indicates which amino acid is now in that position (in this case the Leucine is replace with an Arginine).
The L452R mutation is a mutation that has emerged quite frequently. For example, this mutation was seen in the Epsilon variant (the variant that emerged in California; B1.427/429) which spread throughout the US until late 2021 alongside Delta, which also carries this mutation. This mutation also apparently emerged from a mink population in Denmark (B.1.1.298), although it was relatively short-lived.
Using the information above, we can look at this mutation and infer what we would expect to occur.
This mutation is from a nonpolar amino acid Leucine to a positively charged, hydrogen bond competent amino acid Arginine. As such, we may expect that these new interactions may confer some greater fitness to the virus.
In one study by Motozono, et. al.4, researchers found that the L452R mutation led to great infectivity and fusogenicity of the virus, likely aiding in the virus’ overall fitness.

One suggestion for this increased fitness is an interaction between this new positively charged Arginine and various negatively charged amino acids on the ACEII receptor:
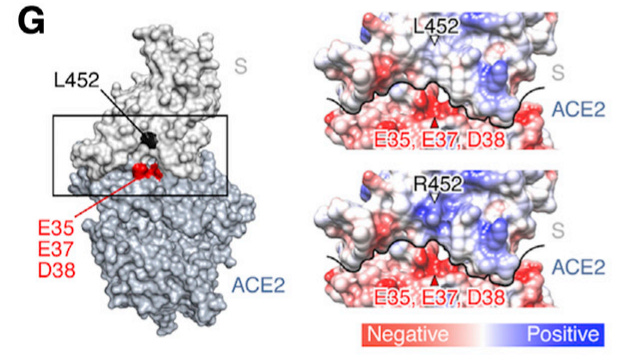
The researchers note this in their discussion:
In the present study, we demonstrate that at least two naturally occurring mutations in the SARS-CoV-2 RBM, L452R and Y453F, escape HLA-restricted cellular immunity and further promote affinity toward the viral receptor ACE2. We also demonstrate that the L452R mutation increases the stability of the S protein and viral infectivity and thereby enhances viral replication. Our data suggest that the L452R mutant escapes HLAA24-restricted cellular immunity and further strengthens its infectivity. Consistent with our findings, Deng et al. performed a pseudovirus assay and showed that the L452R mutation increases viral infectivity (Deng et al., 2021). However, the mechanism of action was not revealed. Here, we demonstrated that the L452R mutation increases viral fusogenicity.
Similar sentiments were provided in a short study by Zhang, et. al.5:
Compared with parental virus and previous variants, the Omicron variant is characterized by decreased hospitalization rates and less severe disease in patients.2 However, how the Omicron variant reduces pathogenicity remains unclear. The Omicron variant has diminished fusogenicity, although the underlying mechanism is unknown. Fusogenicity was shown to be associated with pathogenicity in SARS-CoV-2 patients.3 The L452R mutation, one of the most frequent mutations (Fig. 1a, b), is the only RBD domain mutation that emerges in the Delta variant but is absent in the Omicron variant (Fig. (Fig.1c).1c). It has been reported that L452R mutation increases SARS-CoV-2 fusogenicity and infectivity.4 Here, we developed an L452R mutated Omicron variant (Omicron-L452R) and found that the Omicron-L452R variant rescued fusogenicity and strengthened the high infectivity by enhancing the cleavage of the spike protein. Notably, Omicron-L452R greatly enhanced the ability of Omicron to infect lung tissues of humanized ACE2 mice. Furthermore, the Omicron-L452R variant dramatically enhanced glycolysis in host cells. Our data suggest that the decreased fusogenicity of the Omicron variant is due to a lack of the L452R mutation present in the Delta variant.
It’s interesting that this additional mutation has likely made Ba.4 and Ba.5 behave similar to Delta. Remember that Delta was considered to be more virulent compared to prior strains, and thus this does raise a few concerns as to whether this mutation may make Ba.4/Ba.5 infection more severe.
What’s important for the layperson may be how this affects prior immunity. As can be seen in the mutational lineage above, the L452R mutation occurs in the Receptor Binding Domain (RBD) of the spike protein. More specifically, it’s part of the Receptor Binding Motif (RBM). The RBM is the actual point of contact between the spike protein and the ACEII receptor.
Usually, these regions serve as double-edged swords: if a virus mutates to gain a fitness advantage, this mutation may carry this strain of the virus forward and allow it to propagate further. However, if a mutation arises that reduces binding affinity, this strain would likely be less fit and thus eventually die out.
Remember that the story of variants is a story told by the victor. We only hear of variants with greater fitness because those that have mutated to be less fit will die off, never to be heard from again.
With that being said, most antibodies target the RBD of the spike protein and elicits neutralizing capabilities. However, if the sequence in the RBD mutates antibodies are likely to lose their binding abilities. Generally speaking, mutations in the RBD are usually the main reason why antibodies no longer bind or neutralize a virus. On that note, we can infer that the placement of this mutation is likely to play a role in reduced neutralization.
However, considering that this mutation was also seen in both Epsilon and Delta, it does raise questions as to whether people infected during this timeframe actually have antibodies that target epitopes containing the L452R mutation, which would suggest that these people may be more protected against Ba.4 and Ba.5 relative to other individuals.
This possibility is made very unclear, mostly because many people are unaware of which variant they are infected with. Paired with lack of information on the longevity and breadth of natural immunity we are somewhat left in the dark as to exactly what antibodies and B/T cell responses would hold up against Ba.4/Ba.5. This is just another problem among a host of other problems where we are left to speculate about possibilities due to lack of investigation by the scientific community.
The F486V mutation
This mutation is pretty interesting. It’s a mutation from a nonpolar Phenylalanine (F) to a nonpolar Valine (V). Generally speaking, this mutation that keeps within similar side chain groups may not confer much in the ways of enhanced/reduced pathogenicity. One factor that may be important is the bulky phenyl group that is part of Phenylalanine, and the loss of that group may alleviate some steric hindrance in the protein. However, even with that said general such a slight variation is usually not one to raise many alarms.
In one study by Tsai, et. al.6 researchers used in silico (computer) modeling to see the effects of various RBD mutations on antibody binding.
For F486 they modeled an antibody called MR-17 (an antibody derived from a llama) that interacts with this amino acid (along with other amino acids) to see if mutations within the spike protein would reduce binding to this antibody.

Interestingly, modeling suggests that the sidechain of F is not responsible for interactions with the antibody, but that the oxygen atom from the amide backbone of F may be responsible for interacting with R residues on the antibody.

Heatmap analyses generally showed that any mutation away from Phenylalanine to other amino acids led to reduced binding stability to the M17 antibody. These results are rather strange, considering that for various heatmaps provided even mutations towards a Tyrosine or Tryptophan residue suggests that these mutations are unfavorable for binding to antibodies. All but one heatmap (Foldx) showed reduced antibody association while the heatmap from Foldx showed an increased association if Phenylalanine was changed to Valine. All of this is likely a consequence of in silico analyses which are likely to produce highly variable results due to whichever variables are added to the computational algorithm. Or, it could be due to the more malleable nature of the M17 antibody as compared to the ACEII receptor.
Keep in mind that this study only provides a very narrow window into the role of this F486 mutation and immunity escape. It doesn’t tell us what interactions may occur with human antibodies, and whether this slight change may alter anything significantly.
In short, this mutation is likely to be the least of the concerns here, since the F486V mutation is not one to drastically change the behavior of the spike protein. More information would be needed to sort out if this mutation may be responsible for a host of possible problems down the line.
The Q493 Reversion
Reversions are likely to be a rare occurrence. Usually a mutation would confer some greater fitness onto the virus, and the idea that a dominant subvariant contains a reversion means that more is likely to be at play.
But in order to answer why a reversion may have occurred, we should see what fitness the Q493R mutation provided.
The Q493R mutation is a shift from a Glutamine (polar, uncharged side chain) to an Arginine (polar, positively charged side chain). From there, we may posit that a change to an Arginine with the positive charge may provide fitness to interact with negatively charged residues on the ACEII receptor. This amino acid is located within the RBM, so it is likely to play a critical role in binding to ACEII.
What’s interesting about this mutation is that the Q493R mutation is one of the sole reasons many of the antibodies in prior use generally saw loss of binding activity.
This mutation is responsible for the loss of functionality for Eli Lily’s dual therapy of Bamlanivimab and Etesevimab. Although R is a charged residue, the evidence suggests that the longer side chain length may have led to steric hindrance (too crowded) which may have affected hydrogen bonding7:
The mutation Q493R can induce the disappearance of hydrogen bonds or the collision of antibody CDRH3 region by causing the change of amino acid spatial structure, which may explain the neutralization failure of Etesevimab (class 1/group A) and Bamlanivimab (class 2/group C).
In fact, there is plenty of evidence within the literature to suggest that monoclonal therapy in immunocompromised individuals may lead to selection for a Q493 mutation.
For example, the CDC cites8 the emergence of the Q493R mutation in a 73-year old cancer patient who was given an infusion of Bamlanivimab/Etesivimab. In another case9 an immunocompromised man treated for persistent COVID was provided several doses of Remdesivir and was eventually provided Regeneron on day 143 of his infection. Sequencing analysis indicated the emergence of a Q493K mutation, and was likely to have emerged through persistent infection.
Note that both of the mutations above were mutations towards a positively charged amino acid. This would indicate that this mutation likely favored interactions with negatively charged amino acid residues of the ACEII receptor.
Now, this does raise questions as to whether there may be some association between mAB use and the emergence of these mutations, but keep in mind that mABs are isolated from people who have been naturally infected by COVID, and therefore these antibodies are ones that our bodies are likely to produce ourselves. Essentially, this means that ALL of us- either naturally immune or vaccinated, may select for this mutation.
Interestingly, this reversion back to the Q493 residue carries with it an interesting conundrum. For those of us who were infected with the original Omicron, may we expect that we do not have proper antibodies to target epitopes that contain this amino acid? Would those who were originally infected with variants such as the Wuhan strain, Alpha, or even Delta be protected since they likely have antibodies against this epitope?
William Haseltine from the Forbes article suggests that prior monoclonals may work against Ba.4 and Ba.5, however the existence of other mutations may halt that possibility (Focosi, et. al.):
E484, F490, Q493, and S494 are the 4 aa residues within the spike protein receptor-binding motif that are known to be critical for bamlanivimab binding. Q493 is also among the many more receptor-binding motif residues crucial for interactions with etesivimab.
To provide additional context, Bamlanivimab first loss effectiveness during the emergence of *Beta and Gamma, which caused Eli Lily to withdraw their EUA approval and return later on with the addition of Etesivimab. Interestingly, Bamlanivimab/Etesivimab suffer from the cardinal sin in monoclonal therapy- your antibodies should not share epitopes. Essentially, both Bamlanivimab and Etesivimab share similar epitopes on the RBD of the spike protein, and as soon as a mutation such as the Q493 emerged both lost effectiveness. Granted, Bamlanivimab lost effectiveness around *Beta and Gamma, which raises question as to why Bamlanivimab was included along with Etesivimab in the first place.
*Correction: Alpha was correctly replaced with Beta and Gamma in the paragraph above.
What this would mean is that Etesivimab may show some promise against Ba.4/Ba.5 while Bamlanivimab may still be considered ineffective. This may also suggest that Regeneron may be considered effective in tackling Ba.4 and Ba.5. Further research would be needed in order to figure out which epitopes Regeneron targets, but this may be a time for many medical professionals to reconsider utilizing these monoclonal antibodies once again in the wake of Ba.4/Ba.5 due to this reversion in mutation.
For more on Monoclonal Antibodies in the wake of Omicron, I suggest my Monoclonal Anthology Series.
With all that being said, we need to make sense of the existence of this reversion.
It appears we can make a case that the Q493R mutation may have provided greater affinity to the ACEII receptor. One computer model10 suggests that a Q493K mutation adds additional hydrogen bonding capabilities. Therefore, something similar may arise with Arginine. In general, this would suggest that a reversion back to a Q493 residue should reduce some binding affinity of the spike protein.
So if this reversion is detrimental, why did it come about?
It’s likely that the L452R mutation is aiding the virus the most in its virulence. It could be that the overall net fitness would favor a variant that carried both the L452R mutation as well as the Q493 reversion. Having those two around may be better than not having the L452R mutation present in general.
However, there is a possibility that the 69-70 deletion may have worked here to provide some compensatory mechanism for the reversion. Essentially, the deletion may have come about in response to the Q493 reversion.
This is a bit of a speculation since the emergence of Q493R with Ba.1 came with this deletion. However, a combination of the other mutations may have led to a favoring of the 69-70 deletion, such as the L452R mutation, which actually fits more closely to the results from the Meng, et. al., and may be the primary reason for the 69-70 deletion along with Q493 reversion.
Overall, this reversion is one of the strangest aspects of Ba.4/Ba.5. It likely offers no greater fitness, and actually raises questions as to what this means for prior immunity, or even monoclonal antibody treatments that were revoked as soon as Omicron gained dominance.
The complexities of variants
Ba.4/Ba.5 present with an interesting scenario. Two of the mutations are ones we have seen before (69-70 deletion and L452R) while one is a reversion back to old strains (Q493).
However, in this context there are serving as add-ons (add-offs for Q493?) to Ba.1, which already presented with several dozen mutations of its own. Although we are left wondering how natural immunity or the vaccines will interact with Ba.4/Ba.5, it’s the combination of these mutations within Omicron that makes for a complex scenario.
And it’s the complex scenario that we must make sure to keep in mind. As of now, plenty of conspiracies are running about as to this subvariant, with a few suggesting that this is a variant of the vaccinated, even if most of these assumptions aren’t derived from the literature. It’s easy to look at case rates and make assumptions, but in order to examine the nuances and complexities to suggest that this is a variant of the vaccinated, we need to examine all of the factors required to make such an assumption.
Since this article has gone on too long, the next article will provide a review into immunity variables and whether we can extrapolate anything meaningful to make suppositions, or if a lot of the current assumptions are up in the air (for the time being).
If you enjoyed this post and other works please consider supporting me through a paid Substack subscription or through my Ko-fi. Any bit helps, and it encourages independent creators and journalists outside the mainstream.

Hachmann, N. P., Miller, J., Collier, A. Y., Ventura, J. D., Yu, J., Rowe, M., Bondzie, E. A., Powers, O., Surve, N., Hall, K., & Barouch, D. H. (2022). Neutralization Escape by SARS-CoV-2 Omicron Subvariants BA.2.12.1, BA.4, and BA.5. The New England journal of medicine, 10.1056/NEJMc2206576. Advance online publication. https://doi.org/10.1056/NEJMc2206576
Like with everything named in the sciences, the letter notation for all of these amino acids are extremely hectic. It’s assumed that the amino acids were generally shorthanded based on their first letter. For example, Alanine gets an A and Leucine gets an L. However, there are cases where the first letter overlaps such as alanine and aspartic acid. In these cases, it’s assumed that these amino acids (as well as a few others) receive their letter based on phonetics, such that Aspartic Acid may have a “D” sound and thus gets a D notation while Arginine makes an “R” sound and thus gets designated with an R. Same with Phenylalanine and “F”. Afterwards, the leftover amino acids are assumed to get last dibs and are shortened based on whatever letters were available, which is possibly why Lysine gets a K and Tryptophan gets a W. Keep in mind that this may not actually be how this naming feature came about, but it was how I was taught it during my biochemistry courses.
Meng B, Kemp SA, Papa G, Datir R, Ferreira IATM, Marelli S, Harvey WT, Lytras S, Mohamed A, Gallo G, Thakur N, Collier DA, Mlcochova P; COVID-19 Genomics UK (COG-UK) Consortium, Duncan LM, Carabelli AM, Kenyon JC, Lever AM, De Marco A, Saliba C, Culap K, Cameroni E, Matheson NJ, Piccoli L, Corti D, James LC, Robertson DL, Bailey D, Gupta RK. Recurrent emergence of SARS-CoV-2 spike deletion H69/V70 and its role in the Alpha variant B.1.1.7. Cell Rep. 2021 Jun 29;35(13):109292. doi: 10.1016/j.celrep.2021.109292. Epub 2021 Jun 8. PMID: 34166617; PMCID: PMC8185188.
Motozono C, Toyoda M, Zahradnik J, Saito A, Nasser H, Tan TS, Ngare I, Kimura I, Uriu K, Kosugi Y, Yue Y, Shimizu R, Ito J, Torii S, Yonekawa A, Shimono N, Nagasaki Y, Minami R, Toya T, Sekiya N, Fukuhara T, Matsuura Y, Schreiber G; Genotype to Phenotype Japan (G2P-Japan) Consortium, Ikeda T, Nakagawa S, Ueno T, Sato K. SARS-CoV-2 spike L452R variant evades cellular immunity and increases infectivity. Cell Host Microbe. 2021 Jul 14;29(7):1124-1136.e11. doi: 10.1016/j.chom.2021.06.006. Epub 2021 Jun 15. PMID: 34171266; PMCID: PMC8205251.
Zhang Y, Zhang T, Fang Y, Liu J, Ye Q, Ding L. SARS-CoV-2 spike L452R mutation increases Omicron variant fusogenicity and infectivity as well as host glycolysis. Signal Transduct Target Ther. 2022 Mar 9;7(1):76. doi: 10.1038/s41392-022-00941-z. PMID: 35264568; PMCID: PMC8905570.
Tsai KC, Lee YC, Tseng TS. Comprehensive Deep Mutational Scanning Reveals the Immune-Escaping Hotspots of SARS-CoV-2 Receptor-Binding Domain Targeting Neutralizing Antibodies. Front Microbiol. 2021 Jul 15;12:698365. doi: 10.3389/fmicb.2021.698365. PMID: 34335530; PMCID: PMC8319916.
Li, M., Lou, F. & Fan, H. SARS-CoV-2 variant Omicron: currently the most complete “escapee” from neutralization by antibodies and vaccines. Sig Transduct Target Ther 7, 28 (2022). https://doi.org/10.1038/s41392-022-00880-9
Focosi, D., Novazzi, F., Genoni, A., Dentali, F., Gasperina, D., Baj, A....Maggi, F. (2021). Emergence of SARS-COV-2 Spike Protein Escape Mutation Q493R after Treatment for COVID-19. Emerging Infectious Diseases, 27(10), 2728-2731. https://doi.org/10.3201/eid2710.211538.
Choi B, Choudhary MC, Regan J, Sparks JA, Padera RF, Qiu X, Solomon IH, Kuo HH, Boucau J, Bowman K, Adhikari UD, Winkler ML, Mueller AA, Hsu TY, Desjardins M, Baden LR, Chan BT, Walker BD, Lichterfeld M, Brigl M, Kwon DS, Kanjilal S, Richardson ET, Jonsson AH, Alter G, Barczak AK, Hanage WP, Yu XG, Gaiha GD, Seaman MS, Cernadas M, Li JZ. Persistence and Evolution of SARS-CoV-2 in an Immunocompromised Host. N Engl J Med. 2020 Dec 3;383(23):2291-2293. doi: 10.1056/NEJMc2031364. Epub 2020 Nov 11. PMID: 33176080; PMCID: PMC7673303.
Lupala, C. S., Ye, Y., Chen, H., Su, X. D., & Liu, H. (2022). Mutations on RBD of SARS-CoV-2 Omicron variant result in stronger binding to human ACE2 receptor. Biochemical and biophysical research communications, 590, 34–41. https://doi.org/10.1016/j.bbrc.2021.12.079
Thank you for this, I really appreciate your approach ➡️-"It’s easy to look at case rates and make assumptions, but in order to examine the nuances and complexities to suggest that this is a variant of the vaccinated, we need to examine all of the factors required to make such an assumption."
Excellent work.
Anxiously awaiting the next.