


The Curious Emergence of Omicron
Part I: Introduction and Examining Phylogenetic Trees

It’s been over two years since the first reports of a strange “pneumonia-like illness” began to sprout from Wuhan China. Eventually erupting into a global pandemic, many questions were raised about the origins of the virus. The first reports considered a wet market serial passage through animals such as pangolins. However, the presence of a level 4 biosafety lab nearby the wet market, with evidence of gain-of-function research being conducted, with evidence of experimentation on bat coronaviruses, and with the evidence of bat coronaviruses infecting coal miners several years back being collected and stored, all cast doubt on this “wet market” hypothesis.
Even still, we have no clear evidence of where exactly SARS-COV2 came from.
And then comes the sudden emergence of Omicron. Prior SARS-COV2 variants, although abundant in their mutations, at least exhibited predictable mutations. These variants also tended to follow a similar route of higher infectivity with somewhat reduced virulence (although Delta seems to be both more infectious and virulent).
But Omicron is different; preliminary evidence suggests it’s far more infectious than prior variants and is likely to have an R0 nearly double that of Delta. But it is, in nearly most circumstances, presenting as mild disease. In fact, it’s considered to be the on par with seasonal colds and flus in presentation (there are still those who will suffer severe disease, but it appears those numbers are reduced with Omicron).
Even more strangely, it appears to escape any prior immunity, whether it be from natural infection or prior vaccination regimens, attributed to the “unusually high” number of spike protein mutations. This effect is being seen directly with respect to monoclonal antibody therapy, with most monoclonals in use losing effectiveness aside from Sotrovimab.
Overall, Omicron has presented itself as its own unique beast unlike any of the prior variants we have experienced, which begs the questions as to why Omicron is so different than prior variants, leading to the main concern as to where Omicron may have come from.
Because of how unique Omicron is presenting, we will look at the available research to see what makes Omicron so unique, and if there is any evidence of where it could have come from.
Omicron’s Questionable Lineage
Initial genetic sequencing of Omicron immediately pointed to unique mutations. Not only were there a high number of spike mutations, but there were evidence of mutations unique to prior variants, such as the 69-70 codon deletions which were found in the Alpha variant. As an ex-COVID tester, it was the main way that we were able to notice a difference in variant prevalence when we began testing in the Fall of 2020. The deletion of these codons led to an “S gene drop” during PCR amplification. Therefore, if the S gene did not amplify but the other genes did we used this as a gauge for the presence of Alpha.
The same rules may be used in the case of Omicron, which strangely contains this same deletion and “S gene drop”. What’s even more strange is that this mutation did not occur in any of the other variants. If Omicron were to be derived from prior mutations this would, in some sense, question what variant was Omicron’s closest “ancestor”.
Indeed, this has led to a large search for the closest “relative” to Omicron through the use of a phylogenetic tree. Phylogenetic trees serve as a branching evolutionary map, tying together relationships between different species of animals, bacteria, plants or strains of viruses through comparisons of their genomes. Usually, similarities in genetic profile are used as an indicator of distal/proximal relationships, with more shared genetics suggesting closer relationships.
Using this logic, researchers Kandeel et. al. found that Omicron shared the closest genetic profile with Alpha (emphasis mine):
In comparison to other variants, the number of nucleotide changes in the Omicron genome was in the following order: SARS-CoV-2 USA isolate > Mu variant > Beta variant > Delta variant > Gamma variant > Alpha variant > Omicron variant, with 141, 140, 138, 132, 130, and 109 mutations, respectively (Figure 1). The Alpha variant had the greatest identity rate with Omicron variant (99.63%), followed by Gamma and Mu variants (99.56%). The SARS-CoV-2 USA isolate has the lowest identity (99.53%). Furthermore, Omicron variant showed the greatest number of gaps during genome alignment with other viruses, ranging from 43 to 63 gaps (Figure 2).

The authors suggest that this may be evidence of Omicron having circulated within the population until it was discovered and emerged. However, the likelihood of this should be called into question. With many countries engaging in genomic surveillance it seems difficult to suggest that Omicron remained undetected for well over a year, considering that the Alpha variant emerged several months into the pandemic. This also doesn’t line up with the sudden emergence and rapid transmissibility of this strain of the virus. Considering that many countries have Omicron taking over as the dominant variant nearly two months after the earliest discovery, once again, calls into question how such a highly transmissible virus remained undetected until it took over much of the world.
It’s also worth noting that the viral genome used as a model for Omicron was derived from a sample collected from an infected patient in Botswana, and therefore it should be taken into consideration that the phylogenetic trees being modeled may be biased due to the use of one sample’s viral genome.
So what happens when you scour the database for all iterations of Omicron? Well, the phylogenetic tree comes out a bit different.
In a study by Kannan et. al. researchers utilized a database called the Global Initiative on Sharing Avian Influenza Data, or abbreviated to GISAID, to collect information on possible Omicron variations. GISAID was first started as a global initiative for researchers and scientists to gather global genomic data on the flu. However, the wake of the COVID pandemic has led GISAID to also collect genomic data on SARS-COV2, making it one of the largest databases for COVID genetic information.
Here, researches found 77 different Omicron genomic sequences uploaded to the GISAID database. By narrowing the sequences down to mutations that appear in more than 50% of those 77 genomic sequences, researchers found a total of 30 prevalent spike protein mutations, with 23 of them unique to Omicron, meaning that prior variants did not contain these mutations.
Then, 10 of the Omicron sequences with high quality and high sequence coverage were compared to the 10 most recent sequences from each variant that were uploaded. Using a software program to align the different sequences, the researchers suggested that Omicron was more closely related to Gamma rather than Alpha.
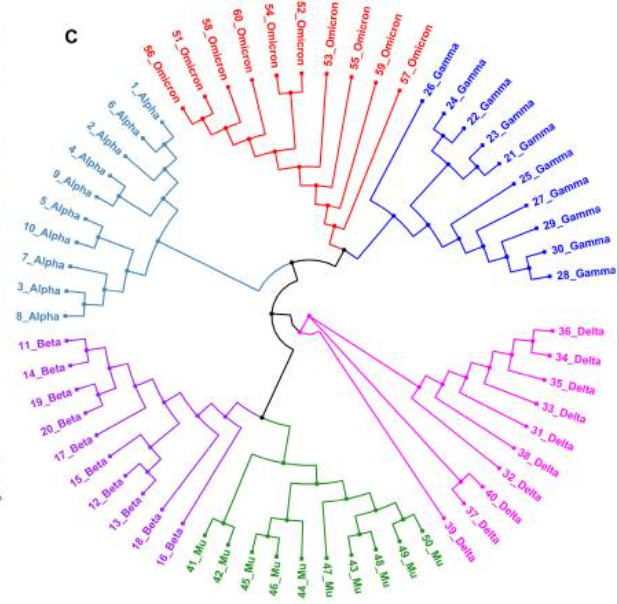
There’s a bit of a disconnect here. One phylogenetic tree suggests Omicron having a closer relationship to Alpha while another says that Gamma may be more closely related. It’s important to point out that both Alpha and Gamma are considered to be closely related, and that even with differences in mutations an overwhelming portion of SARS-COV2’s genome remains conserved across variants.
The differences in phylogenetic trees are likely due to sample bias. The Kandeel et. al. study uses genomic data collected from the earliest discovered sample from Botswana. It’s important to remember that this is the earliest discovered Omicron sample, not the first emergence of the variant, which does not take into account any mutations that may have arisen beforehand.
The Kannan et. al. study uses evidence from consistent, prevalent mutations as the basis for its phylogenetic tree. However, if the concern with the Kandeel et. al. study is that mutations may occur in the timeframe between Omicron’s emergence and its discovery, the utilization of a database is likely to utilize data that is consistent with the most widespread variant, rather than the earliest variant. Considering that GISAID collects global data, it’s hard not to argue that there’s a good chance of viral/viral or viral/host influence on creating a variety of Omicron variations that gain prevalence in the global population. In that regard, the measure of mutation prevalence is not indicative of the progenitor form of Omicron more than the most widely circulating mutations.
However this still doesn’t take into account the strange disappearance and reemergence of the 69-70 spike protein deletion. Considering that all of the other variants of concern that have circulated over the past 2 years (aside from Omicron) do not contain this deletion it seems strange to see its sudden reemergence. Granted, if more infectious variants arise they could mask the appearance of this deletion, although viral evolution would still fall under selective pressure; if this variant is less infectious it would stand to reason that it would eventually fall out of circulation while other variants dominant.
This leads me and my amateur opinion to lean more towards Alpha being the closest “relative” to Omicron, mostly on account of the 69-70 spike protein deletion. If we take into account the prevalence of Alpha and Gamma, we can also exclude the possibility of Gamma on the basis that Omicron is suggested to have emerged from Botswana/South Africa, where Gamma does not seem to have seen the same level of circulation that Alpha appears to have.
Either way, the evidence suggests Omicron shares ancestry with variants that emerged early on in the pandemic. To share such a relationship with older variants begs the question as to where Omicron may have been, and how it could have suddenly emerged to dominate the globe; all questions we may hope to piece together with cumulative evidence.
Part II will examine evidence of drastic dynamic changes in Omicron and how that changes the viral landscape.
Hmmm, curiouser and curiouser....